当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Proteomics Goes to Court: A Statistical Foundation for Forensic Toxin/Organism Identification Using Bottom-Up Proteomics
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-08-28 , DOI: 10.1021/acs.jproteome.8b00212 Kristin H. Jarman 1 , Natalie C. Heller 1 , Sarah C. Jenson 2 , Janine R. Hutchison 2 , Brooke L. Deatherage Kaiser 2 , Samuel H. Payne 3 , David S. Wunschel 2 , Eric D. Merkley 2
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-08-28 , DOI: 10.1021/acs.jproteome.8b00212 Kristin H. Jarman 1 , Natalie C. Heller 1 , Sarah C. Jenson 2 , Janine R. Hutchison 2 , Brooke L. Deatherage Kaiser 2 , Samuel H. Payne 3 , David S. Wunschel 2 , Eric D. Merkley 2
Affiliation
|
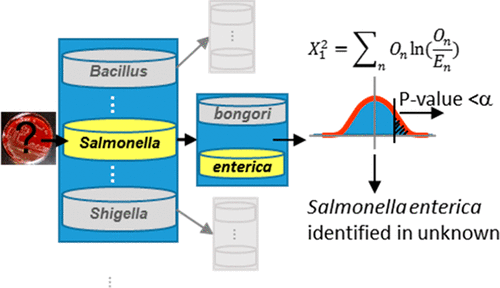
Bottom-up proteomics is increasingly being used to characterize unknown environmental, clinical, and forensic samples. Proteomics-based bacterial identification typically proceeds by tabulating peptide “hits” (i.e., confidently identified peptides) associated with the organisms in a database; those organisms with enough hits are declared present in the sample. This approach has proven to be successful in laboratory studies; however, important research gaps remain. First, the common-practice reliance on unique peptides for identification is susceptible to a phenomenon known as signal erosion. Second, no general guidelines are available for determining how many hits are needed to make a confident identification. These gaps inhibit the transition of this approach to real-world forensic samples where conditions vary and large databases may be needed. In this work, we propose statistical criteria that overcome the problem of signal erosion and can be applied regardless of the sample quality or data analysis pipeline. These criteria are straightforward, producing a p-value on the result of an organism or toxin identification. We test the proposed criteria on 919 LC-MS/MS data sets originating from 2 toxins and 32 bacterial strains acquired using multiple data collection platforms. Results reveal a > 95% correct species-level identification rate, demonstrating the effectiveness and robustness of proteomics-based organism/toxin identification.
中文翻译:

蛋白质组学上法庭:使用自下而上的蛋白质组学鉴定法医毒素/有机物的统计基础
自下而上的蛋白质组学被越来越多地用于表征未知的环境,临床和法医样品。基于蛋白质组学的细菌鉴定通常通过在数据库中将与生物体相关的肽“命中”(即可靠鉴定的肽)制成表格来进行;那些具有足够命中率的生物被宣布存在于样品中。实践证明,这种方法在实验室研究中是成功的。但是,重要的研究差距仍然存在。首先,通常的做法是依靠独特的肽段进行识别,这种现象容易受到信号侵蚀的影响。其次,没有通用的准则可用于确定需要多少次点击才能进行可靠的识别。这些差距阻碍了这种方法向实际法医样品的过渡,在这种情况下,条件不断变化,可能需要大型数据库。在这项工作中,我们提出了克服信号腐蚀问题的统计标准,无论样本质量或数据分析流程如何,都可以应用统计标准。这些标准很简单,产生了有机物或毒素鉴定结果的p值。我们在919 LC-MS / MS数据集上测试了提议的标准,该数据集来自使用多种数据收集平台获得的2种毒素和32种细菌菌株。结果表明,正确的物种水平识别率> 95%,证明了基于蛋白质组学的生物/毒素识别的有效性和鲁棒性。
更新日期:2018-08-29
中文翻译:
蛋白质组学上法庭:使用自下而上的蛋白质组学鉴定法医毒素/有机物的统计基础
自下而上的蛋白质组学被越来越多地用于表征未知的环境,临床和法医样品。基于蛋白质组学的细菌鉴定通常通过在数据库中将与生物体相关的肽“命中”(即可靠鉴定的肽)制成表格来进行;那些具有足够命中率的生物被宣布存在于样品中。实践证明,这种方法在实验室研究中是成功的。但是,重要的研究差距仍然存在。首先,通常的做法是依靠独特的肽段进行识别,这种现象容易受到信号侵蚀的影响。其次,没有通用的准则可用于确定需要多少次点击才能进行可靠的识别。这些差距阻碍了这种方法向实际法医样品的过渡,在这种情况下,条件不断变化,可能需要大型数据库。在这项工作中,我们提出了克服信号腐蚀问题的统计标准,无论样本质量或数据分析流程如何,都可以应用统计标准。这些标准很简单,产生了有机物或毒素鉴定结果的p值。我们在919 LC-MS / MS数据集上测试了提议的标准,该数据集来自使用多种数据收集平台获得的2种毒素和32种细菌菌株。结果表明,正确的物种水平识别率> 95%,证明了基于蛋白质组学的生物/毒素识别的有效性和鲁棒性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号