当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Using graphs to quantify energetic and structural order in semicrystalline oligothiophene thin films†
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2018-07-26 00:00:00 , DOI: 10.1039/c8me00028j Ellen Van 1, 2, 3, 4 , Matthew Jones 4, 5, 6, 7 , Eric Jankowski 4, 5, 6, 7 , Olga Wodo 2, 3, 4, 8
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2018-07-26 00:00:00 , DOI: 10.1039/c8me00028j Ellen Van 1, 2, 3, 4 , Matthew Jones 4, 5, 6, 7 , Eric Jankowski 4, 5, 6, 7 , Olga Wodo 2, 3, 4, 8
Affiliation
|
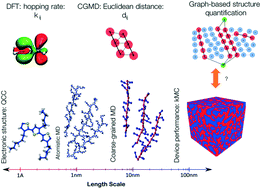
In semicrystalline conjugated polymer thin films, the mobility of charges depends on the arrangement of the individual polymer chains. In particular, the ordering of the polymer backbones affects the charge transport within the film, as electron transfer generally occurs along the backbones with alternating single and double bonds. In this paper, we demonstrate that polymer ordering should be discussed not only in terms of structural but also energetic ordering of polymer chains. We couple data from molecular dynamics simulations and quantum chemical calculations to quantify both structural and energetic ordering of polymer chains. We leverage a graph-based representation of the polymer chains to quantify the transport pathways in a computationally efficient way. Next, we formulate the morphological descriptors that correlate well with hole mobility determined using kinetic Monte Carlo simulations. We show that the shortest and fastest path calculations are predictive of mobility in equilibrated morphologies. In this sense, we leverage graph-based descriptors to provide a basis for the quantitative structure property relationships.
中文翻译:

使用图表量化半结晶低聚噻吩薄膜的能级和结构顺序†
在半结晶共轭聚合物薄膜中,电荷的迁移率取决于各个聚合物链的排列。特别地,聚合物电子主链的排序影响膜内的电荷传输,因为电子转移通常沿着具有交替的单键和双键的主链发生。在本文中,我们证明了不仅应该在结构上而且应该在聚合物链的能量有序方面讨论聚合物的有序性。我们将分子动力学模拟和量子化学计算中的数据耦合在一起,以量化聚合物链的结构和能量顺序。我们利用聚合物链的基于图形的表示形式,以计算有效的方式量化运输途径。下一个,我们制定了与动力学蒙特卡洛模拟确定的空穴迁移率很好相关的形态学描述子。我们显示最短和最快的路径计算可预测平衡形态中的移动性。从这个意义上讲,我们利用基于图的描述符为定量结构特性关系提供基础。
更新日期:2018-07-26
中文翻译:
使用图表量化半结晶低聚噻吩薄膜的能级和结构顺序†
在半结晶共轭聚合物薄膜中,电荷的迁移率取决于各个聚合物链的排列。特别地,聚合物电子主链的排序影响膜内的电荷传输,因为电子转移通常沿着具有交替的单键和双键的主链发生。在本文中,我们证明了不仅应该在结构上而且应该在聚合物链的能量有序方面讨论聚合物的有序性。我们将分子动力学模拟和量子化学计算中的数据耦合在一起,以量化聚合物链的结构和能量顺序。我们利用聚合物链的基于图形的表示形式,以计算有效的方式量化运输途径。下一个,我们制定了与动力学蒙特卡洛模拟确定的空穴迁移率很好相关的形态学描述子。我们显示最短和最快的路径计算可预测平衡形态中的移动性。从这个意义上讲,我们利用基于图的描述符为定量结构特性关系提供基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号