当前位置:
X-MOL 学术
›
Macromolecules
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Conformational and Dynamic Properties of Poly(ethylene oxide) in BMIM+BF4–: A Microsecond Computer Simulation Study Using ab Initio Force Fields
Macromolecules ( IF 5.1 ) Pub Date : 2018-07-11 00:00:00 , DOI: 10.1021/acs.macromol.8b01002 Chang Yun Son 1 , Jesse G. McDaniel 1 , Qiang Cui 1 , Arun Yethiraj 1
Macromolecules ( IF 5.1 ) Pub Date : 2018-07-11 00:00:00 , DOI: 10.1021/acs.macromol.8b01002 Chang Yun Son 1 , Jesse G. McDaniel 1 , Qiang Cui 1 , Arun Yethiraj 1
Affiliation
|
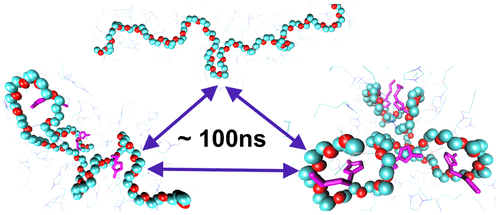
The behavior of polymers in complex solvents is interesting from a fundamental perspective and of practical importance from the standpoint of polymer processing. There has been recent interest in the conformational and dynamic properties of polymer in room temperature ionic liquids, with conflicting predictions from computations using models with different resolutions and conflicting results of experiments from different groups. In this work, we develop a first-principles, nonpolarizable united atom (UA) force field for a mixture of poly(ethylene oxide) (PEO) in the ionic liquid BMIM+BF4–. The UA force field is benchmarked against ab initio calculations, and the PEO atomic charges are parametrized to implicitly capture the polarization contribution to the solvation energy of a single PEO molecule in BMIM+BF4–. The UA model allows one to perform multi-microsecond molecular dynamics simulations. This is necessary because the conformational relaxation correlation times are of the order of 100 ns. The simulations predict that the radius of gyration, Rg, scales with molecular weight, Rg ∼ Mwν with ν ≈ 0.56 in the temperature range 300–600 K, consistent with experiment, seemingly in between a self-avoiding walk and an ideal chain. An examination of the snapshots of the polymer demonstrates, however, that the polymer conformations are composed of ringlike and linear segments, with ringlike parts of the chain wrapped around cations of the ionic liquid. The slow dynamics arises from the barrier to unwrapping the ringlike segments of the polymer. The mean-square displacement shows three regimes which we interpret as confinement, Zimm, and diffusive. The simulations emphasize the importance of accurate force fields and microsecond simulations in obtaining reliable results for polymers and elucidate important correlation effects for polymers in strongly interacting solvents.
中文翻译:

BMIM + BF 4 –中聚环氧乙烷的构象和动力学性质:使用从头算力场的微秒计算机模拟研究
从基本的角度看,聚合物在复杂溶剂中的行为是有趣的,从聚合物加工的角度看,它在实际中具有重要意义。近年来,人们对室温离子液体中聚合物的构象和动力学性质产生了兴趣,使用具有不同分辨率的模型进行的计算得出的预测相互矛盾,并且来自不同组的实验得出的结论相互矛盾。在这项工作中,我们为离子液体BMIM + BF 4中的聚环氧乙烷(PEO)混合物开发了第一性原理,不可极化的联合原子(UA)力场–。UA力场相对于从头算计算得到基准,并且对PEO原子电荷进行了参数设置,以隐式捕获极化对BMIM + BF 4 –中单个PEO分子的溶剂化能的贡献。UA模型允许执行多微秒分子动力学模拟。这是必需的,因为构象弛豫相关时间约为100 ns。该模拟预测到回转,半径ř克,分子量秤,- [R克〜中号瓦特ν在300-600 K的温度范围内ν≈0.56,这与实验一致,似乎在自我规避的行走和理想的链之间。然而,对聚合物快照的检查表明,聚合物构象由环状和线性链段组成,链的环状部分包裹在离子液体的阳离子周围。缓慢的动力学起因于打开聚合物的环状链段的屏障。均方位移显示了三种状态,我们将其解释为限制,Zimm和扩散。模拟强调精确力场和微秒模拟对于获得聚合物可靠结果的重要性,并阐明在强相互作用溶剂中聚合物的重要关联效应。
更新日期:2018-07-11
中文翻译:
BMIM + BF 4 –中聚环氧乙烷的构象和动力学性质:使用从头算力场的微秒计算机模拟研究
从基本的角度看,聚合物在复杂溶剂中的行为是有趣的,从聚合物加工的角度看,它在实际中具有重要意义。近年来,人们对室温离子液体中聚合物的构象和动力学性质产生了兴趣,使用具有不同分辨率的模型进行的计算得出的预测相互矛盾,并且来自不同组的实验得出的结论相互矛盾。在这项工作中,我们为离子液体BMIM + BF 4中的聚环氧乙烷(PEO)混合物开发了第一性原理,不可极化的联合原子(UA)力场–。UA力场相对于从头算计算得到基准,并且对PEO原子电荷进行了参数设置,以隐式捕获极化对BMIM + BF 4 –中单个PEO分子的溶剂化能的贡献。UA模型允许执行多微秒分子动力学模拟。这是必需的,因为构象弛豫相关时间约为100 ns。该模拟预测到回转,半径ř克,分子量秤,- [R克〜中号瓦特ν在300-600 K的温度范围内ν≈0.56,这与实验一致,似乎在自我规避的行走和理想的链之间。然而,对聚合物快照的检查表明,聚合物构象由环状和线性链段组成,链的环状部分包裹在离子液体的阳离子周围。缓慢的动力学起因于打开聚合物的环状链段的屏障。均方位移显示了三种状态,我们将其解释为限制,Zimm和扩散。模拟强调精确力场和微秒模拟对于获得聚合物可靠结果的重要性,并阐明在强相互作用溶剂中聚合物的重要关联效应。










































 京公网安备 11010802027423号
京公网安备 11010802027423号