当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Repulsion–dispersion parameters for the modelling of organic molecular crystals containing N, O, S and Cl
Faraday Discussions ( IF 3.4 ) Pub Date : 2018-07-12 , DOI: 10.1039/c8fd00064f Christina A. Gatsiou 1, 2, 3, 4, 5 , Claire S. Adjiman 1, 2, 3, 4, 5 , Constantinos C. Pantelides 1, 2, 3, 4, 5
Faraday Discussions ( IF 3.4 ) Pub Date : 2018-07-12 , DOI: 10.1039/c8fd00064f Christina A. Gatsiou 1, 2, 3, 4, 5 , Claire S. Adjiman 1, 2, 3, 4, 5 , Constantinos C. Pantelides 1, 2, 3, 4, 5
Affiliation
|
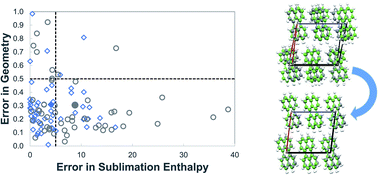
In lattice energy models that combine ab initio and empirical components, it is important to ensure consistency between these components so that meaningful quantitative results are obtained. A method for deriving parameters of atom–atom repulsion dispersion potentials for crystals, tailored to different ab initio models, is presented. It is based on minimization of the sum of squared deviations between experimental and calculated structures and energies. The solution algorithm is designed to avoid convergence to local minima in the parameter space by combining a deterministic low-discrepancy sequence for the generation of multiple initial parameter guesses with an efficient local minimization algorithm. The proposed approach is applied to derive transferable exp-6 potential parameters suitable for use in conjunction with a distributed multipole electrostatics model derived from isolated molecule charge densities calculated at the M06/6-31G(d,p) level of theory. Data for hydrocarbons, azahydrocarbons, oxohydrocarbons, organosulphur compounds and chlorohydrocarbons are used for the estimation. A good fit is achieved for the new set of parameters with a mean absolute error in sublimation enthalpies of 4.1 kJ mol−1 and an average rmsd15 of 0.31 Å. The parameters are found to perform well on a separate cross-validation set of 39 compounds.
中文翻译:

排斥-扩散参数,用于建模包含N,O,S和Cl的有机分子晶体
在结合了从头算起和经验成分的晶格能量模型中,重要的是要确保这些成分之间的一致性,以便获得有意义的定量结果。根据不同的从头算来推导晶体的原子-原子斥力弥散势参数的方法模型,介绍。它基于最小化实验结构和计算结构与能量之间平方差的总和。通过将用于生成多个初始参数猜测值的确定性低差异序列与有效的局部最小化算法相结合,可将求解算法设计为避免在参数空间中收敛至局部最小值。所提出的方法适用于推导适用于与分布式多极静电模型结合使用的可转移exp-6电位参数,该模型从理论上M06 / 6-31G(d,p)级别计算出的孤立分子电荷密度推导而来。估计使用碳氢化合物,氮杂碳氢化合物,氧代碳氢化合物,有机硫化合物和氯代烃的数据。-1,平均rmsd 15为0.31Å。发现该参数在单独的39种化合物的交叉验证组中表现良好。
更新日期:2018-10-26
中文翻译:
排斥-扩散参数,用于建模包含N,O,S和Cl的有机分子晶体
在结合了从头算起和经验成分的晶格能量模型中,重要的是要确保这些成分之间的一致性,以便获得有意义的定量结果。根据不同的从头算来推导晶体的原子-原子斥力弥散势参数的方法模型,介绍。它基于最小化实验结构和计算结构与能量之间平方差的总和。通过将用于生成多个初始参数猜测值的确定性低差异序列与有效的局部最小化算法相结合,可将求解算法设计为避免在参数空间中收敛至局部最小值。所提出的方法适用于推导适用于与分布式多极静电模型结合使用的可转移exp-6电位参数,该模型从理论上M06 / 6-31G(d,p)级别计算出的孤立分子电荷密度推导而来。估计使用碳氢化合物,氮杂碳氢化合物,氧代碳氢化合物,有机硫化合物和氯代烃的数据。-1,平均rmsd 15为0.31Å。发现该参数在单独的39种化合物的交叉验证组中表现良好。


























 京公网安备 11010802027423号
京公网安备 11010802027423号