当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Steered Molecular Dynamics Simulation in Rational Drug Design
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-07-05 00:00:00 , DOI: 10.1021/acs.jcim.8b00261 Phuc-Chau Do 1 , Eric H. Lee 2 , Ly Le 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-07-05 00:00:00 , DOI: 10.1021/acs.jcim.8b00261 Phuc-Chau Do 1 , Eric H. Lee 2 , Ly Le 1
Affiliation
|
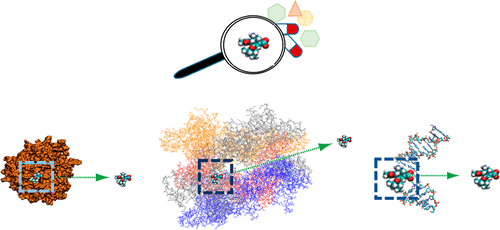
Conventional de novo drug design is time consuming, laborious, and resource intensive. In recent years, emerging in silico approaches have been proven to be critical to accelerate the process of bringing drugs to market. Molecular dynamics (MD) simulations of single molecule and molecular complexes have been commonly applied to achieve accurate binding modes and binding energies of drug-receptor interactions. A derivative of MD, namely, steered molecular dynamics (SMD), has been demonstrated as a promising tool for rational drug design. In this paper, we review various studies over the last 20 years using SMD simulations, thus paving the way to determine the relationship between protein structure and function. In addition, the paper highlights the use of SMD simulation for in silico drug design. We also aim to establish an understanding on the key interactions which play a crucial role in the stabilization of peptide–ligand interfaces, the binding and unbinding mechanism of the ligand–protein complex, the mechanism of ligand translocating via membrane, and the ranking of different ligands on receptors as therapeutic candidates.
中文翻译:

合理药物设计中的分子动力学模拟
常规的从头药物设计是费时,费力且资源密集的。近年来,事实证明,新兴的计算机方法对于加快将药物推向市场的过程至关重要。单分子和分子复合物的分子动力学(MD)模拟已普遍用于实现精确的结合模式和药物-受体相互作用的结合能。MD的衍生产品,即操纵分子动力学(SMD),已被证明是进行合理药物设计的有前途的工具。在本文中,我们回顾了过去20年使用SMD模拟进行的各种研究,从而为确定蛋白质结构与功能之间的关系铺平了道路。此外,本文重点介绍了SMD仿真在以下方面的用途:在硅药物设计中。我们还旨在建立对关键相互作用的理解,这些相互作用在肽-配体界面的稳定,配体-蛋白质复合物的结合和非结合机制,配体通过膜转运的机制以及不同分子的排名中起着至关重要的作用。受体上的配体作为治疗候选物。
更新日期:2018-07-05
中文翻译:
合理药物设计中的分子动力学模拟
常规的从头药物设计是费时,费力且资源密集的。近年来,事实证明,新兴的计算机方法对于加快将药物推向市场的过程至关重要。单分子和分子复合物的分子动力学(MD)模拟已普遍用于实现精确的结合模式和药物-受体相互作用的结合能。MD的衍生产品,即操纵分子动力学(SMD),已被证明是进行合理药物设计的有前途的工具。在本文中,我们回顾了过去20年使用SMD模拟进行的各种研究,从而为确定蛋白质结构与功能之间的关系铺平了道路。此外,本文重点介绍了SMD仿真在以下方面的用途:在硅药物设计中。我们还旨在建立对关键相互作用的理解,这些相互作用在肽-配体界面的稳定,配体-蛋白质复合物的结合和非结合机制,配体通过膜转运的机制以及不同分子的排名中起着至关重要的作用。受体上的配体作为治疗候选物。








































 京公网安备 11010802027423号
京公网安备 11010802027423号