Chemical Physics ( IF 2.0 ) Pub Date : 2018-06-26 , DOI: 10.1016/j.chemphys.2018.06.017 M.P. Taylor , G.A. Worth
|
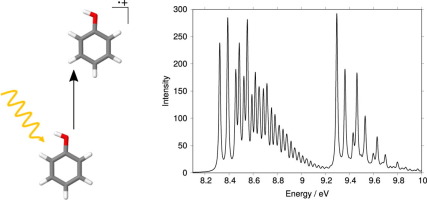
The photoelectron spectrum for the two lowest ionisation bands of phenol has been simulated using quantum dynamic methods. A vibronic coupling Hamiltonian was set up consisting of seven vibrational modes and the first two ionised states. Parameters for the model are obtained by fitting adiabatic surfaces to a series of points calculated using ab initio methods. Such a model allows non-adiabatic couplings between the states to be included. CASSCF calculations used in this work provide reliable quantum chemical information for the model and the calculated photoelectron spectrum shows good agreement to experiment. The vibrational fine structure of both bands are reassessed and different assignments to those previously reported are detailed. The existence of a conical intersection between the ionised states is reported and its role in the dynamics of phenol upon ionisation is examined.
中文翻译:
振动耦合模型计算苯酚的光电子能谱
使用量子动力学方法模拟了苯酚的两个最低电离带的光电子能谱。建立了由七个振动模式和前两个电离状态组成的振动耦合哈密顿量。通过将绝热表面拟合到使用从头算起的一系列点来获得模型的参数方法。这样的模型允许包括状态之间的非绝热耦合。这项工作中使用的CASSCF计算为模型提供了可靠的量子化学信息,并且计算出的光电子光谱与实验具有很好的一致性。重新评估了两个频段的振动精细结构,并详细说明了与先前报告的不同的分配。报道了电离态之间存在圆锥形相交,并研究了其在电离后苯酚动力学中的作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号