当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine learning for predicting product distributions in catalytic regioselective reactions†
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2018-06-22 00:00:00 , DOI: 10.1039/c8cp03141j Sayan Banerjee 1, 2, 3, 4 , A. Sreenithya 1, 2, 3, 4 , Raghavan B. Sunoj 1, 2, 3, 4
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2018-06-22 00:00:00 , DOI: 10.1039/c8cp03141j Sayan Banerjee 1, 2, 3, 4 , A. Sreenithya 1, 2, 3, 4 , Raghavan B. Sunoj 1, 2, 3, 4
Affiliation
|
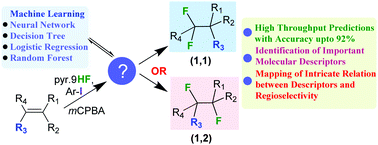
Gaining predictable control over various forms of selectivities, such as enantio- and/or regio-selectivities, has been a long-standing goal in chemical catalysis. Although a number of factors such as the molecular features of the reactants and catalysts, as well as the reaction conditions, can influence the outcome of a reaction, it is not quite conspicuous as to what combinations of these parameters would offer a desired form of selectivity. We use machine learning tools, such as the neural network (NN), decision tree (DT), logistic regression (LR) and Random forest algorithms, to (a) analyze the outcome of an important catalytic regio-selective difluorination reaction of alkenes, and (b) decipher the complex interplay of various molecular parameters and their non-linear dependencies. The connection between what features of alkenes will yield 1,1-difluorination and how subtle changes would steer the reaction to 1,2-difluorination under identical conditions is enunciated. The NN was able to accurately predict whether a given alkene would yield a 1,1- or 1,2-difluorinated product. A combination of DT and the random forest classifier offered important chemical insights, which could be used in making a more rational choice of the reactant alkene for the desired regioisomeric product. The results could have far reaching implications in predicting which regioisomer is likely to be formed under a given set of conditions, and thus this technique is capable of expediting the development of catalytic transformations.
中文翻译:

机器学习可预测催化区域选择性反应中的产物分布†
对各种形式的选择性(例如对映体)获得可预测的控制-和/或区域选择性一直是化学催化的一个长期目标。尽管许多因素(例如反应物和催化剂的分子特征以及反应条件)会影响反应的结果,但是这些参数的哪种组合会提供所需的选择性形式并不十分明显。 。我们使用机器学习工具,例如神经网络(NN),决策树(DT),逻辑回归(LR)和随机森林算法,来(a)分析烯烃重要的催化区域选择性二氟化反应的结果, (b)解释各种分子参数及其非线性依赖性的复杂相互作用。烯烃的哪些特征将产生1,1-二氟与微妙的变化将反应引导至1,阐明在相同条件下的2-二氟化。NN能够准确预测给定的烯烃会产生1,1或1,2-二氟代产物。DT和随机森林分类器的组合提供了重要的化学见解,可用于为所需的区域异构体产品更合理地选择反应物烯烃。该结果在预测在给定条件下可能形成哪种区域异构体时可能具有深远的意义,因此该技术能够加快催化转化的发展。可以用于更合理地选择所需的区域异构体产物的反应物烯烃。该结果在预测在给定条件下可能形成哪种区域异构体时可能具有深远的意义,因此该技术能够加快催化转化的发展。可以用于更合理地选择所需的区域异构体产物的反应物烯烃。该结果在预测在给定条件下可能形成哪种区域异构体时可能具有深远的意义,因此该技术能够加快催化转化的发展。
更新日期:2018-06-22
中文翻译:
机器学习可预测催化区域选择性反应中的产物分布†
对各种形式的选择性(例如对映体)获得可预测的控制-和/或区域选择性一直是化学催化的一个长期目标。尽管许多因素(例如反应物和催化剂的分子特征以及反应条件)会影响反应的结果,但是这些参数的哪种组合会提供所需的选择性形式并不十分明显。 。我们使用机器学习工具,例如神经网络(NN),决策树(DT),逻辑回归(LR)和随机森林算法,来(a)分析烯烃重要的催化区域选择性二氟化反应的结果, (b)解释各种分子参数及其非线性依赖性的复杂相互作用。烯烃的哪些特征将产生1,1-二氟与微妙的变化将反应引导至1,阐明在相同条件下的2-二氟化。NN能够准确预测给定的烯烃会产生1,1或1,2-二氟代产物。DT和随机森林分类器的组合提供了重要的化学见解,可用于为所需的区域异构体产品更合理地选择反应物烯烃。该结果在预测在给定条件下可能形成哪种区域异构体时可能具有深远的意义,因此该技术能够加快催化转化的发展。可以用于更合理地选择所需的区域异构体产物的反应物烯烃。该结果在预测在给定条件下可能形成哪种区域异构体时可能具有深远的意义,因此该技术能够加快催化转化的发展。可以用于更合理地选择所需的区域异构体产物的反应物烯烃。该结果在预测在给定条件下可能形成哪种区域异构体时可能具有深远的意义,因此该技术能够加快催化转化的发展。


























 京公网安备 11010802027423号
京公网安备 11010802027423号