当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Configurational-Bias Monte Carlo Back-Mapping Algorithm for Efficient and Rapid Conversion of Coarse-Grained Water Structures into Atomistic Models
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-07-05 , DOI: 10.1021/acs.jpcb.8b01791 Troy D. Loeffler , Henry Chan , Badri Narayanan , Mathew J. Cherukara , Stephen Gray , Subramanian K. R. S. Sankaranarayanan
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-07-05 , DOI: 10.1021/acs.jpcb.8b01791 Troy D. Loeffler , Henry Chan , Badri Narayanan , Mathew J. Cherukara , Stephen Gray , Subramanian K. R. S. Sankaranarayanan
|
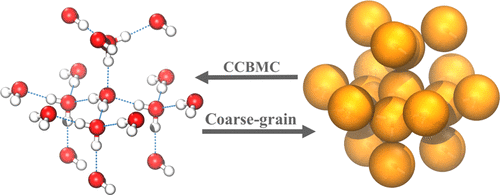
Coarse-grained molecular dynamics (MD) simulations represent a powerful approach to simulate longer time scale and larger length scale phenomena than those accessible to all-atom models. The gain in efficiency, however, comes at the cost of atomistic details. The reverse transformation, also known as back mapping, of coarse-grained beads into their atomistic constituents represents a major challenge. Most existing approaches are limited to specific molecules or specific force fields and often rely on running a long-time atomistic MD of the back-mapped configuration to arrive at an optimal solution. Such approaches are problematic when dealing with systems with high diffusion barriers. Here, we introduce a new extension of the configurational-bias Monte Carlo (CBMC) algorithm, which we term the crystalline-configurational-bias Monte Carlo (C-CBMC) algorithm, which allows rapid and efficient conversion of a coarse-grained model back into its atomistic representation. Although the method is generic, we use a coarse-grained water model as a representative example and demonstrate the back mapping or reverse transformation for model systems ranging from the ice–liquid water interface to amorphous and crystalline ice configurations. A series of simulations using the TIP4P/Ice model are performed to compare the new CBMC method to several other standard Monte Carlo and molecular dynamics-based back-mapping techniques. In all of the cases, the C-CBMC algorithm is able to find optimal hydrogen-bonded configuration many thousand evaluations/steps sooner than the other methods compared within this paper. For crystalline ice structures, such as a hexagonal, cubic, and cubic-hexagonal stacking disorder structures, the C-CBMC was able to find structures that were between 0.05 and 0.1 eV/water molecule lower in energy than the ground-state energies predicted by the other methods. Detailed analysis of the atomistic structures shows a significantly better global hydrogen positioning when contrasted with the existing simpler back-mapping methods. The errors in the radial distribution functions (RDFs) of back-mapped configuration relative to reference configuration for the C-CBMC, MD, and MC were found to be 6.9, 8.7, and 12.9, respectively, for the hexagonal system. For the cubic system, the relative errors of the RDFs for the C-CBMC, MD, and MC were found to be 18.2, 34.6, and 39.0, respectively. Our results demonstrate the efficiency and efficacy of our new back-mapping approach, especially for crystalline systems where simple force-field-based relaxations have a tendency to get trapped in local minima.
中文翻译:

有效地将粗粒水结构快速有效地转换为原子模型的配置偏向蒙特卡洛反映射算法
粗粒度分子动力学(MD)模拟代表了一种比所有原子模型可访问的模拟更长的时间尺度和更大的长度尺度现象的强大方法。然而,效率的提高是以牺牲原子细节为代价的。粗粒珠粒向其原子成分的反向转化(也称为反向映射)是一个重大挑战。大多数现有方法仅限于特定的分子或特定的力场,并且通常依赖于运行后映射配置的长时间原子MD来获得最佳解决方案。当处理具有高扩散障碍的系统时,这样的方法是有问题的。在这里,我们介绍了配置偏向蒙特卡洛(CBMC)算法的新扩展,我们将其称为“结晶构型偏向蒙特卡罗(C-CBMC)”算法,该算法可将粗粒度模型快速有效地转换回其原子表示。尽管该方法是通用的,但我们使用粗粒水模型作为代表示例,并演示了从冰–液态水界面到无定形和结晶冰构型的模型系统的反向映射或反向转换。执行了一系列使用TIP4P / Ice模型的仿真,以将新的CBMC方法与其他几种标准的蒙特卡洛和基于分子动力学的反映射技术进行比较。在所有情况下,与本文中比较的其他方法相比,C-CBMC算法能够更快地找到数千个评估/步骤的最佳氢键构型。对于晶体冰结构,例如六方,立方和立方六方堆积无序结构,C-CBMC能够找到能量比水分子预测的基态能量低0.05至0.1 eV /水分子的结构。其他方法。与现有的更简单的反向映射方法相比,对原子结构的详细分析显示出明显更好的全局氢定位。对于六边形系统,相对于C-CBMC,MD和MC的参考配置,后映射配置的径向分布函数(RDF)的误差分别为6.9、8.7和12.9。对于三次系统,发现C-CBMC,MD和MC的RDF的相对误差分别为18.2、34.6和39.0。
更新日期:2018-07-08
中文翻译:
有效地将粗粒水结构快速有效地转换为原子模型的配置偏向蒙特卡洛反映射算法
粗粒度分子动力学(MD)模拟代表了一种比所有原子模型可访问的模拟更长的时间尺度和更大的长度尺度现象的强大方法。然而,效率的提高是以牺牲原子细节为代价的。粗粒珠粒向其原子成分的反向转化(也称为反向映射)是一个重大挑战。大多数现有方法仅限于特定的分子或特定的力场,并且通常依赖于运行后映射配置的长时间原子MD来获得最佳解决方案。当处理具有高扩散障碍的系统时,这样的方法是有问题的。在这里,我们介绍了配置偏向蒙特卡洛(CBMC)算法的新扩展,我们将其称为“结晶构型偏向蒙特卡罗(C-CBMC)”算法,该算法可将粗粒度模型快速有效地转换回其原子表示。尽管该方法是通用的,但我们使用粗粒水模型作为代表示例,并演示了从冰–液态水界面到无定形和结晶冰构型的模型系统的反向映射或反向转换。执行了一系列使用TIP4P / Ice模型的仿真,以将新的CBMC方法与其他几种标准的蒙特卡洛和基于分子动力学的反映射技术进行比较。在所有情况下,与本文中比较的其他方法相比,C-CBMC算法能够更快地找到数千个评估/步骤的最佳氢键构型。对于晶体冰结构,例如六方,立方和立方六方堆积无序结构,C-CBMC能够找到能量比水分子预测的基态能量低0.05至0.1 eV /水分子的结构。其他方法。与现有的更简单的反向映射方法相比,对原子结构的详细分析显示出明显更好的全局氢定位。对于六边形系统,相对于C-CBMC,MD和MC的参考配置,后映射配置的径向分布函数(RDF)的误差分别为6.9、8.7和12.9。对于三次系统,发现C-CBMC,MD和MC的RDF的相对误差分别为18.2、34.6和39.0。










































 京公网安备 11010802027423号
京公网安备 11010802027423号