当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Simulations of Interfacial Tension of Liquid–Liquid Ternary Mixtures Using Optimized Parametrization for Coarse-Grained Models
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-06-15 00:00:00 , DOI: 10.1021/acs.jctc.8b00357 David Steinmetz 1 , Benoit Creton 1 , Véronique Lachet 1, 2 , Bernard Rousseau 2 , Carlos Nieto-Draghi 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-06-15 00:00:00 , DOI: 10.1021/acs.jctc.8b00357 David Steinmetz 1 , Benoit Creton 1 , Véronique Lachet 1, 2 , Bernard Rousseau 2 , Carlos Nieto-Draghi 1
Affiliation
|
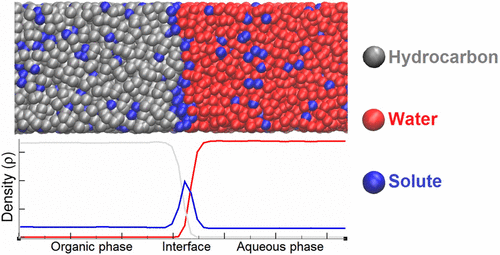
In this work, liquid–liquid systems are studied by means of coarse-grained Monte Carlo simulations (CG-MC) and Dissipative Particle Dynamics (DPD). A methodology is proposed to reproduce liquid–liquid equilibrium (LLE) and to provide variation of interfacial tension (IFT), as a function of the solute concentration. A key step is the parametrization method based on the use of the Flory–Huggins parameter between DPD beads to calculate solute/solvent interactions. Parameters are determined using a set of experimental compositional data of LLE, following four different approaches. These approaches are evaluated, and the results obtained are compared to analyze advantages/disadvantages of each one. These methodologies have been compared through their application on six systems: water/benzene/1,4-dioxane,water/chloroform/acetone, water/benzene/acetic acid, water/benzene/2-propanol, water/hexane/acetone, and water/hexane/2-propanol. CG-MC simulations in the Gibbs (NVT) ensemble have been used to check the validity of parametrization approaches for LLE reproduction. Then, CG-MC simulations in the osmotic (μsoluteNsolventPzzT) ensemble were carried out considering the two liquid phases with an explicit interface. This step allows one to work at the same bulk concentrations as the experimental data by imposing the precise bulk phase compositions and predicting the interface composition. Finally, DPD simulations were used to predict IFT values for different solute concentrations. Our results on variation of IFT with solute concentration in bulk phases are in good agreement with experimental data, but some deviations can be observed for systems containing hexane molecules.
中文翻译:

粗粒体模型优化参数化对液-液三元混合物界面张力的模拟
在这项工作中,通过粗粒度蒙特卡洛模拟(CG-MC)和耗散粒子动力学(DPD)研究了液-液系统。提出了一种方法来重现液-液平衡(LLE),并提供界面张力(IFT)随溶质浓度变化的变化。关键步骤是基于在DPD珠粒之间使用Flory-Huggins参数来计算溶质/溶剂相互作用的参数化方法。使用四种不同方法,使用一组LLE实验组成数据确定参数。对这些方法进行了评估,并对获得的结果进行了比较,以分析每种方法的优缺点。这些方法已通过在六个系统上的应用进行了比较:水/苯/ 1,4-二恶烷,水/氯仿/丙酮,水/苯/乙酸,水/苯/ 2-丙醇,水/己烷/丙酮和水/己烷/ 2-丙醇。Gibbs(NVT)集成中的CG-MC模拟已用于检查LLE再现的参数化方法的有效性。然后,渗透压的CG-MC模拟(μ考虑到具有明确界面的两种液相,进行了固溶N溶剂( P zz T)集成。通过施加精确的本体相组成并预测界面组成,这一步骤可以使人们在与实验数据相同的本体浓度下工作。最后,DPD模拟用于预测不同溶质浓度的IFT值。我们在IFT中随固相浓度变化的结果与实验数据非常吻合,但是对于包含己烷分子的系统,可以观察到一些偏差。
更新日期:2018-06-15
中文翻译:
粗粒体模型优化参数化对液-液三元混合物界面张力的模拟
在这项工作中,通过粗粒度蒙特卡洛模拟(CG-MC)和耗散粒子动力学(DPD)研究了液-液系统。提出了一种方法来重现液-液平衡(LLE),并提供界面张力(IFT)随溶质浓度变化的变化。关键步骤是基于在DPD珠粒之间使用Flory-Huggins参数来计算溶质/溶剂相互作用的参数化方法。使用四种不同方法,使用一组LLE实验组成数据确定参数。对这些方法进行了评估,并对获得的结果进行了比较,以分析每种方法的优缺点。这些方法已通过在六个系统上的应用进行了比较:水/苯/ 1,4-二恶烷,水/氯仿/丙酮,水/苯/乙酸,水/苯/ 2-丙醇,水/己烷/丙酮和水/己烷/ 2-丙醇。Gibbs(NVT)集成中的CG-MC模拟已用于检查LLE再现的参数化方法的有效性。然后,渗透压的CG-MC模拟(μ考虑到具有明确界面的两种液相,进行了固溶N溶剂( P zz T)集成。通过施加精确的本体相组成并预测界面组成,这一步骤可以使人们在与实验数据相同的本体浓度下工作。最后,DPD模拟用于预测不同溶质浓度的IFT值。我们在IFT中随固相浓度变化的结果与实验数据非常吻合,但是对于包含己烷分子的系统,可以观察到一些偏差。


























 京公网安备 11010802027423号
京公网安备 11010802027423号