Journal of Catalysis ( IF 6.5 ) Pub Date : 2018-06-14 , DOI: 10.1016/j.jcat.2018.05.016 Stephanie Kwon , Prashant Deshlahra , Enrique Iglesia
|
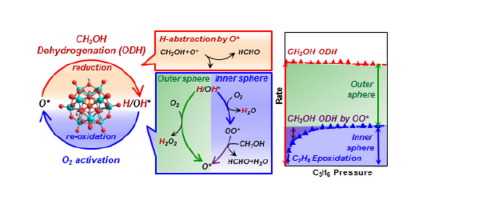
Catalytic redox cycles involve dioxygen activation via peroxo (OO∗) or H2O2 species, denoted as inner-sphere and outer-sphere routes respectively, for metal-oxo catalysts solvated by liquids. On solid oxides, O2 activation is typically more facile than the reduction part of redox cycles, making kinetic inquiries difficult at steady-state. These steps are examined here for oxidative alkanol dehydrogenation (ODH) by scavenging OO∗ species with C3H6 to form epoxides and by energies and barriers from density functional theory. Alkanols react with O-atoms (O∗) in oxides to form vicinal OH pairs that eliminate H2O to form OO∗ at O-vacancies formed or react with O2 to give H2O2. OO∗ reacts with alkanols to re-form O∗ via steps favored over OO∗ migrations, otherwise required to oxidize non-vicinal vacancies. C3H6 epoxidizes by reaction with OO∗ with rates that increase with C3H6 pressure, but reach constant values as all OO∗ species react with C3H6 at high C3H6/alkanol ratios. Asymptotic epoxidation/ODH rate ratios are smaller than unity, because outer-sphere routes that shuttle O-atoms via H2O2(g) are favored over endoergic vacancy formation required for inner-sphere routes. The relative contributions of these two routes are influenced by H2O, because vacancies, required to form OO∗, react with H2O to form OH pairs and H2O2. OO∗-mediated routes and epoxidation become favored at low coverages of reduced centers, prevalent for less reactive alkanols and lower alkanol/O2 ratios, because H2O2 then reacts preferentially with O∗ (forming OO∗), instead of vacancies (forming O∗/H2O). Such kinetic shunts between two routes compensate for lower barriers required to form H2O2 than OO∗. These re-oxidation routes prefer molecular donor (H2O2) or acceptor (alkanol) to perform stepwise two-electron oxidations by dioxygen, instead of kinetically demanding O-atom migrations. The quantitative descriptions, derived from theory and experiment on Mo-based polyoxometalate clusters with known structures, bring together the dioxygen chemistry in liquid-phase oxidations, including electro-catalysis and monooxygenase enzymes, and oxide surfaces into a common framework, while suggesting a practical process for epoxidation by kinetically coupling with ODH reaction.
中文翻译:
金属氧化物催化的Mars-van Krevelen氧化还原循环中的双氧活化途径
催化氧化还原循环涉及通过过氧化物(OO *)或H 2 O 2物种的双氧活化,分别被液体溶剂化的金属-氧代催化剂表示为内球和外球路线。在固态氧化物上,O 2活化通常比氧化还原循环的还原部分更容易,从而使稳态下的动力学查询变得困难。在此检查这些步骤的氧化性链烷醇脱氢(ODH),方法是用C 3 H 6清除OO *物质以形成环氧化物,并通过密度泛函理论的能量和势垒进行检查。烷醇与氧化物中的O原子(O **)反应形成邻位OH对,从而消除H 2O在形成的空位处形成OO *或与O 2反应生成H 2 O 2。OO *与烷醇反应,通过比OO *迁移更有利的步骤重新形成O *,否则需要氧化非邻位空位。C 3 H 6通过与OO *反应而环氧化,其速率随C 3 H 6的压力而增加,但随着所有OO *种类在高C 3 H 6下与C 3 H 6反应而达到恒定值。/链烷醇比。渐进环氧化/ ODH速率比小于1,因为通过O 2原子通过H 2 O 2(g)穿梭O原子的外球途径比内球途径所需的内能空位形成更受青睐。这两种途径的相对贡献受H 2 O影响,因为形成OO *所需的空位与H 2 O反应形成OH对和H 2 O 2。OO *介导的途径和环氧化反应在低覆盖率的还原中心反应中变得尤为重要,这主要是由于反应性较低的链烷醇和较低的链烷醇/ O 2比,因为H 2 O 2然后优先与O *(形成OO *)反应,而不是与空位(形成O * / H 2 O)反应。两条路径之间的这种动力学分流补偿了形成H 2 O 2所需的比OO ∗更低的势垒。这些再氧化途径更倾向于分子供体(H 2 O 2)或受体(链烷醇)通过双氧进行逐步的两电子氧化,而不是在动力学上要求O原子迁移。定量描述源于具有已知结构的Mo基多金属氧酸盐簇的理论和实验,将液相氧化中的双氧化学(包括电催化和单加氧酶和氧化物表面)整合到一个通用框架中,同时提出了实用的方法通过与ODH反应动力学偶联进行环氧化的方法。









































 京公网安备 11010802027423号
京公网安备 11010802027423号