当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adlayer Dynamics Drives CO Activation in Ru-Catalyzed Fischer–Tropsch Synthesis
ACS Catalysis ( IF 11.3 ) Pub Date : 2018-06-12 00:00:00 , DOI: 10.1021/acscatal.8b01232 Lucas Foppa 1 , Marcella Iannuzzi 2 , Christophe Copéret 1 , Aleix Comas-Vives 1
ACS Catalysis ( IF 11.3 ) Pub Date : 2018-06-12 00:00:00 , DOI: 10.1021/acscatal.8b01232 Lucas Foppa 1 , Marcella Iannuzzi 2 , Christophe Copéret 1 , Aleix Comas-Vives 1
Affiliation
|
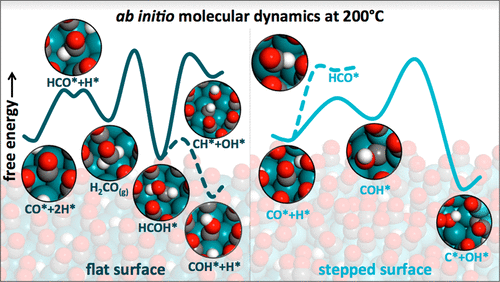
The first step of the Fischer–Tropsch synthesis (FTS) consists of the carbon monoxide activation at ca. 200 °C on catalyst metal surfaces covered with dense adlayers. Based on first-principles calculations, two main mechanisms for the C–O bond cleavage have been proposed for Ru catalysts: the direct and the hydrogen-assisted routes on step-edges and flat surfaces, respectively. However, commonly used static density functional theory (DFT) methods describe FTS adlayers using nonmobile adsorbed CO (CO*) species, while under reaction conditions the adsorbates diffuse and interact with each other. Here, we use ab initio molecular dynamics (AIMD) simulations on Ru flat and stepped model surfaces covered with CO* and H* to interrogate the effect of adlayer dynamics on the preferred reaction mechanisms. We show that hydrogen-assisted CO activation mechanisms via hydroxyl-carbonyl (COH*) intermediates formed on step-edges are the most favored according to AIMD simulations. Therefore, both step-edges and surface hydrogen play a key role in CO cleavage during Ru-catalyzed FTS at high CO* coverage. Direct comparison with static DFT results reveals that the dynamic adlayer significantly affects the relative stability of reaction intermediates and shows that the mobility of adsorbed molecules modulates the reaction paths, calling for a systematic analysis of reaction networks on complex systems at high coverages using AIMD simulations.
中文翻译:

Adlayer Dynamics在Ru催化的Fischer-Tropsch合成中驱动CO活化
费-托合成(FTS)的第一步是由大约一氧化碳活化。200°C在覆盖有致密添加剂的催化剂金属表面上。基于第一性原理计算,已提出了Ru催化剂C-O键断裂的两个主要机理:分别在台阶边缘和平坦表面上的直接和氢辅助途径。但是,常用的静态密度泛函理论(DFT)方法描述了使用非移动吸附的CO(CO *)物质的FTS吸附层,而在反应条件下,吸附物彼此扩散并相互作用。在这里,我们在覆盖有CO *和H *的Ru平坦和阶梯状模型表面上使用从头算分子动力学(AIMD)模拟,以研究添加剂动力学对优选反应机理的影响。我们表明,根据AIMD模拟,通过在步缘上形成的羟基羰基(COH *)中间体的氢辅助CO活化机理是最受青睐的。因此,在高CO *覆盖率的Ru催化FTS期间,台阶边缘和表面氢都在CO裂解中起关键作用。与静态DFT结果的直接比较表明,动态吸附层会显着影响反应中间体的相对稳定性,并表明吸附分子的迁移率可调节反应路径,因此需要使用AIMD模拟对高覆盖率复杂系统上的反应网络进行系统分析。在高CO *覆盖率的Ru催化的FTS中,台阶边缘和表面氢都在CO裂解中起关键作用。与静态DFT结果的直接比较表明,动态吸附层会显着影响反应中间体的相对稳定性,并表明吸附分子的迁移率可调节反应路径,因此需要使用AIMD模拟对高覆盖率复杂系统上的反应网络进行系统分析。在高CO *覆盖率的Ru催化的FTS中,台阶边缘和表面氢都在CO裂解中起关键作用。与静态DFT结果的直接比较表明,动态吸附层会显着影响反应中间体的相对稳定性,并表明吸附分子的迁移性可调节反应路径,因此需要使用AIMD模拟对高覆盖率复杂系统上的反应网络进行系统分析。
更新日期:2018-06-12
中文翻译:
Adlayer Dynamics在Ru催化的Fischer-Tropsch合成中驱动CO活化
费-托合成(FTS)的第一步是由大约一氧化碳活化。200°C在覆盖有致密添加剂的催化剂金属表面上。基于第一性原理计算,已提出了Ru催化剂C-O键断裂的两个主要机理:分别在台阶边缘和平坦表面上的直接和氢辅助途径。但是,常用的静态密度泛函理论(DFT)方法描述了使用非移动吸附的CO(CO *)物质的FTS吸附层,而在反应条件下,吸附物彼此扩散并相互作用。在这里,我们在覆盖有CO *和H *的Ru平坦和阶梯状模型表面上使用从头算分子动力学(AIMD)模拟,以研究添加剂动力学对优选反应机理的影响。我们表明,根据AIMD模拟,通过在步缘上形成的羟基羰基(COH *)中间体的氢辅助CO活化机理是最受青睐的。因此,在高CO *覆盖率的Ru催化FTS期间,台阶边缘和表面氢都在CO裂解中起关键作用。与静态DFT结果的直接比较表明,动态吸附层会显着影响反应中间体的相对稳定性,并表明吸附分子的迁移率可调节反应路径,因此需要使用AIMD模拟对高覆盖率复杂系统上的反应网络进行系统分析。在高CO *覆盖率的Ru催化的FTS中,台阶边缘和表面氢都在CO裂解中起关键作用。与静态DFT结果的直接比较表明,动态吸附层会显着影响反应中间体的相对稳定性,并表明吸附分子的迁移率可调节反应路径,因此需要使用AIMD模拟对高覆盖率复杂系统上的反应网络进行系统分析。在高CO *覆盖率的Ru催化的FTS中,台阶边缘和表面氢都在CO裂解中起关键作用。与静态DFT结果的直接比较表明,动态吸附层会显着影响反应中间体的相对稳定性,并表明吸附分子的迁移性可调节反应路径,因此需要使用AIMD模拟对高覆盖率复杂系统上的反应网络进行系统分析。









































 京公网安备 11010802027423号
京公网安备 11010802027423号