当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crystal Structure Prediction via Deep Learning
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2018-06-06 , DOI: 10.1021/jacs.8b03913 Kevin Ryan 1 , Jeff Lengyel 1 , Michael Shatruk 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2018-06-06 , DOI: 10.1021/jacs.8b03913 Kevin Ryan 1 , Jeff Lengyel 1 , Michael Shatruk 1
Affiliation
|
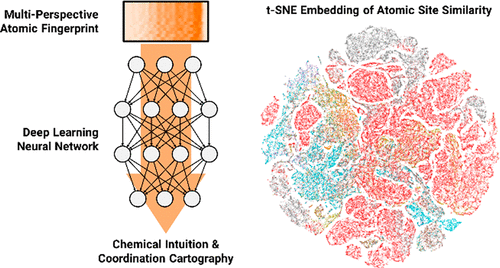
We demonstrate the application of deep neural networks as a machine-learning tool for the analysis of a large collection of crystallographic data contained in the crystal structure repositories. Using input data in the form of multiperspective atomic fingerprints, which describe coordination topology around unique crystallographic sites, we show that the neural-network model can be trained to effectively distinguish chemical elements based on the topology of their crystallographic environment. The model also identifies structurally similar atomic sites in the entire data set of ∼50000 crystal structures, essentially uncovering trends that reflect the periodic table of elements. The trained model was used to analyze templates derived from the known crystal structures in order to predict the likelihood of forming new compounds that could be generated by placing elements into these structural templates in a combinatorial fashion. Statistical analysis of predictive performance of the neural-network model, which was applied to a test set of structures never seen by the model during training, indicates its ability to predict known elemental compositions with a high likelihood of success. In ∼30% of cases, the known compositions were found among the top 10 most likely candidates proposed by the model. These results suggest that the approach developed in this work can be used to effectively guide the synthetic efforts in the discovery of new materials, especially in the case of systems composed of three or more chemical elements.
中文翻译:

通过深度学习进行晶体结构预测
我们展示了深度神经网络作为机器学习工具的应用,用于分析晶体结构库中包含的大量晶体数据。使用多视角原子指纹形式的输入数据,描述了独特晶体学位点周围的协调拓扑结构,我们表明可以训练神经网络模型以根据其晶体学环境的拓扑结构有效区分化学元素。该模型还在大约 50000 个晶体结构的整个数据集中识别了结构相似的原子位点,基本上揭示了反映元素周期表的趋势。训练后的模型用于分析源自已知晶体结构的模板,以预测形成新化合物的可能性,这些化合物可以通过以组合方式将元素放入这些结构模板中而产生。神经网络模型的预测性能的统计分析,应用于模型在训练期间从未见过的结构测试集,表明其预测已知元素组成的能力很有可能成功。在约 30% 的情况下,已知成分位于模型提出的前 10 名最有可能的候选者中。这些结果表明,这项工作中开发的方法可用于有效指导发现新材料的合成工作,
更新日期:2018-06-06
中文翻译:
通过深度学习进行晶体结构预测
我们展示了深度神经网络作为机器学习工具的应用,用于分析晶体结构库中包含的大量晶体数据。使用多视角原子指纹形式的输入数据,描述了独特晶体学位点周围的协调拓扑结构,我们表明可以训练神经网络模型以根据其晶体学环境的拓扑结构有效区分化学元素。该模型还在大约 50000 个晶体结构的整个数据集中识别了结构相似的原子位点,基本上揭示了反映元素周期表的趋势。训练后的模型用于分析源自已知晶体结构的模板,以预测形成新化合物的可能性,这些化合物可以通过以组合方式将元素放入这些结构模板中而产生。神经网络模型的预测性能的统计分析,应用于模型在训练期间从未见过的结构测试集,表明其预测已知元素组成的能力很有可能成功。在约 30% 的情况下,已知成分位于模型提出的前 10 名最有可能的候选者中。这些结果表明,这项工作中开发的方法可用于有效指导发现新材料的合成工作,










































 京公网安备 11010802027423号
京公网安备 11010802027423号