当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-Principles Modeling of Polaron Formation in TiO2 Polymorphs
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-06-06 00:00:00 , DOI: 10.1021/acs.jctc.8b00199 A. R. Elmaslmane 1 , M. B. Watkins 2 , K. P. McKenna 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-06-06 00:00:00 , DOI: 10.1021/acs.jctc.8b00199 A. R. Elmaslmane 1 , M. B. Watkins 2 , K. P. McKenna 1
Affiliation
|
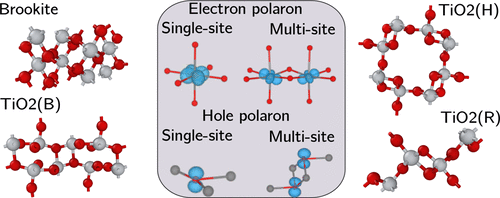
We present a computationally efficient and predictive methodology for modeling the formation and properties of electron and hole polarons in solids. Through a nonempirical and self-consistent optimization of the fraction of Hartree–Fock exchange (α) in a hybrid functional, we ensure the generalized Koopmans’ condition is satisfied and self-interaction error is minimized. The approach is applied to model polaron formation in known stable and metastable phases of TiO2 including anatase, rutile, brookite, TiO2(H), TiO2(R), and TiO2(B). Electron polarons are predicted to form in rutile, TiO2(H), and TiO2(R) (with trapping energies ranging from −0.02 eV to −0.35 eV). In rutile the electron localizes on a single Ti ion, whereas in TiO2(H) and TiO2(R) the electron is distributed across two neighboring Ti sites. Hole polarons are predicted to form in anatase, brookite, TiO2(H), TiO2(R), and TiO2(B) (with trapping energies ranging from −0.16 eV to −0.52 eV). In anatase, brookite, and TiO2(B) holes localize on a single O ion, whereas in TiO2(H) and TiO2(R) holes can also be distributed across two O sites. We find that the optimized α has a degree of transferability across the phases, with α = 0.115 describing all phases well. We also note the approach yields accurate band gaps, with anatase, rutile, and brookite within six percent of experimental values. We conclude our study with a comparison of the alignment of polaron charge transition levels across the different phases. Since the approach we describe is only two to three times more expensive than a standard density functional theory calculation, it is ideally suited to model charge trapping at complex defects (such as surfaces and interfaces) in a range of materials relevant for technological applications but previously inaccessible to predictive modeling.
中文翻译:

TiO 2多晶型物极化子形成的第一性原理模拟
我们提出了一种计算效率高的预测方法,用于对固体中电子和空穴极化子的形成和性质进行建模。通过对混合函数中Hartree-Fock交换(α)的分数进行非经验且自洽的优化,我们确保满足广义Koopmans条件,并将自交互误差最小化。该方法适用于模拟已知的TiO 2稳定和亚稳相中的极化子形成,包括锐钛矿,金红石,板钛矿,TiO 2(H),TiO 2(R)和TiO 2(B)。预计会在金红石,TiO 2(H)和TiO 2中形成电子极化子(R)(俘获能范围为-0.02 eV至-0.35 eV)。在金红石中,电子位于单个Ti离子上,而在TiO 2(H)和TiO 2(R)中,电子分布在两个相邻的Ti位置。空穴极化子预计在锐钛矿,板钛矿,TiO 2(H),TiO 2(R)和TiO 2(B)中形成(俘获能范围为-0.16 eV至-0.52 eV)。在锐钛矿中,板钛矿和TiO 2(B)孔位于单个O离子上,而在TiO 2(H)和TiO 2中(R)孔也可以分布在两个O位置。我们发现优化的α在各个相之间具有一定程度的可传递性,其中α= 0.115很好地描述了所有相。我们还注意到该方法产生了精确的带隙,锐钛矿,金红石和板钛矿在实验值的百分之六以内。我们通过比较不同相中极化子电荷跃迁水平的对准来结束我们的研究。由于我们描述的方法仅比标准密度泛函理论计算贵2至3倍,因此非常适合于建模与技术应用相关的一系列材料中复杂缺陷(例如表面和界面)处的电荷陷阱模型,但是以前预测模型无法访问。
更新日期:2018-06-06
中文翻译:
TiO 2多晶型物极化子形成的第一性原理模拟
我们提出了一种计算效率高的预测方法,用于对固体中电子和空穴极化子的形成和性质进行建模。通过对混合函数中Hartree-Fock交换(α)的分数进行非经验且自洽的优化,我们确保满足广义Koopmans条件,并将自交互误差最小化。该方法适用于模拟已知的TiO 2稳定和亚稳相中的极化子形成,包括锐钛矿,金红石,板钛矿,TiO 2(H),TiO 2(R)和TiO 2(B)。预计会在金红石,TiO 2(H)和TiO 2中形成电子极化子(R)(俘获能范围为-0.02 eV至-0.35 eV)。在金红石中,电子位于单个Ti离子上,而在TiO 2(H)和TiO 2(R)中,电子分布在两个相邻的Ti位置。空穴极化子预计在锐钛矿,板钛矿,TiO 2(H),TiO 2(R)和TiO 2(B)中形成(俘获能范围为-0.16 eV至-0.52 eV)。在锐钛矿中,板钛矿和TiO 2(B)孔位于单个O离子上,而在TiO 2(H)和TiO 2中(R)孔也可以分布在两个O位置。我们发现优化的α在各个相之间具有一定程度的可传递性,其中α= 0.115很好地描述了所有相。我们还注意到该方法产生了精确的带隙,锐钛矿,金红石和板钛矿在实验值的百分之六以内。我们通过比较不同相中极化子电荷跃迁水平的对准来结束我们的研究。由于我们描述的方法仅比标准密度泛函理论计算贵2至3倍,因此非常适合于建模与技术应用相关的一系列材料中复杂缺陷(例如表面和界面)处的电荷陷阱模型,但是以前预测模型无法访问。










































 京公网安备 11010802027423号
京公网安备 11010802027423号