当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-06-21 , DOI: 10.1021/acs.jpcb.8b02144 Matthew Carter Childers 1 , Valerie Daggett 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-06-21 , DOI: 10.1021/acs.jpcb.8b02144 Matthew Carter Childers 1 , Valerie Daggett 1
Affiliation
|
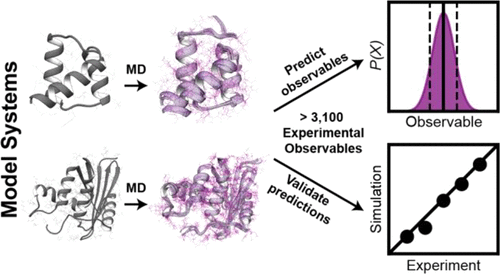
Far from the static, idealized conformations deposited into structural databases, proteins are highly dynamic molecules that undergo conformational changes on temporal and spatial scales that may span several orders of magnitude. These conformational changes, often intimately connected to the functional roles that proteins play, may be obscured by traditional biophysical techniques. Over the past 40 years, molecular dynamics (MD) simulations have complemented these techniques by providing the “hidden” atomistic details that underlie protein dynamics. However, there are limitations of the degree to which molecular simulations accurately and quantitatively describe protein motions. Here we show that although four molecular dynamics simulation packages (AMBER, GROMACS, NAMD, and ilmm) reproduced a variety of experimental observables for two different proteins (engrailed homeodomain and RNase H) equally well overall at room temperature, there were subtle differences in the underlying conformational distributions and the extent of conformational sampling obtained. This leads to ambiguity about which results are correct, as experiment cannot always provide the necessary detailed information to distinguish between the underlying conformational ensembles. However, the results with different packages diverged more when considering larger amplitude motion, for example, the thermal unfolding process and conformational states sampled, with some packages failing to allow the protein to unfold at high temperature or providing results at odds with experiment. While most differences between MD simulations performed with different packages are attributed to the force fields themselves, there are many other factors that influence the outcome, including the water model, algorithms that constrain motion, how atomic interactions are handled, and the simulation ensemble employed. Here four different MD packages were tested each using best practices as established by the developers, utilizing three different protein force fields and three different water models. Differences between the simulated protein behavior using two different packages but the same force field, as well as two different packages with different force fields but the same water models and approaches to restraining motion, show how other factors can influence the behavior, and it is incorrect to place all the blame for deviations and errors on force fields or to expect improvements in force fields alone to solve such problems.
中文翻译:

根据基础构象集合验证针对实验可观察物的分子动力学模拟
蛋白质不是存放在结构数据库中的静态理想化构象,而是高度动态的分子,它们在时空范围内发生可能跨越数个数量级的构象变化。这些构象变化通常与蛋白质发挥的功能作用密切相关,传统的生物物理技术可能会掩盖这些构象变化。在过去的40年中,分子动力学(MD)模拟通过提供构成蛋白质动力学基础的“隐藏”原子细节,对这些技术进行了补充。但是,分子模拟准确和定量地描述蛋白质运动的程度是有限的。在这里,我们展示了四个分子动力学模拟程序包(AMBER,GROMACS,NAMD和il毫米)再现了两种不同蛋白质的实验观察值,在室温下总体上同样好,两种蛋白质的潜在构象分布和获得的构象采样程度均存在细微差异。由于实验无法始终提供必要的详细信息来区分潜在的构象整体,因此这导致对哪个结果正确的认识不清。然而,当考虑更大的振幅运动时,不同包装的结果差异更大,例如,热展开过程和采样的构象状态,有些包装无法使蛋白质在高温下展开或无法提供与实验不一致的结果。尽管使用不同包装进行的MD模拟之间的大多数差异都归因于力场本身,但还有许多其他因素会影响结果,包括水模型,约束运动的算法,原子相互作用的处理方式以及所采用的模拟集合。在这里,使用开发人员建立的最佳实践对四个不同的MD软件包进行了测试,它们使用了三个不同的蛋白质力场和三个不同的水模型。使用两个不同包装但具有相同力场的模拟蛋白质行为之间的差异,以及使用不同作用场但具有相同水模型和约束运动方法的两个不同包装之间的差异表明了其他因素如何影响该行为,
更新日期:2018-06-22
中文翻译:
根据基础构象集合验证针对实验可观察物的分子动力学模拟
蛋白质不是存放在结构数据库中的静态理想化构象,而是高度动态的分子,它们在时空范围内发生可能跨越数个数量级的构象变化。这些构象变化通常与蛋白质发挥的功能作用密切相关,传统的生物物理技术可能会掩盖这些构象变化。在过去的40年中,分子动力学(MD)模拟通过提供构成蛋白质动力学基础的“隐藏”原子细节,对这些技术进行了补充。但是,分子模拟准确和定量地描述蛋白质运动的程度是有限的。在这里,我们展示了四个分子动力学模拟程序包(AMBER,GROMACS,NAMD和il毫米)再现了两种不同蛋白质的实验观察值,在室温下总体上同样好,两种蛋白质的潜在构象分布和获得的构象采样程度均存在细微差异。由于实验无法始终提供必要的详细信息来区分潜在的构象整体,因此这导致对哪个结果正确的认识不清。然而,当考虑更大的振幅运动时,不同包装的结果差异更大,例如,热展开过程和采样的构象状态,有些包装无法使蛋白质在高温下展开或无法提供与实验不一致的结果。尽管使用不同包装进行的MD模拟之间的大多数差异都归因于力场本身,但还有许多其他因素会影响结果,包括水模型,约束运动的算法,原子相互作用的处理方式以及所采用的模拟集合。在这里,使用开发人员建立的最佳实践对四个不同的MD软件包进行了测试,它们使用了三个不同的蛋白质力场和三个不同的水模型。使用两个不同包装但具有相同力场的模拟蛋白质行为之间的差异,以及使用不同作用场但具有相同水模型和约束运动方法的两个不同包装之间的差异表明了其他因素如何影响该行为,










































 京公网安备 11010802027423号
京公网安备 11010802027423号