当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantitative Assessment of Methods Used To Obtain Rate Constants from Molecular Dynamics Simulations—Translocation of Cholesterol across Lipid Bilayers
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-06-04 00:00:00 , DOI: 10.1021/acs.jctc.8b00150 Hugo A. L. Filipe 1, 2 , Matti Javanainen 3, 4 , Armindo Salvador 1, 2 , Adelino M. Galvão 5 , Ilpo Vattulainen 3, 4, 6 , Luís M. S. Loura 1, 7 , Maria João Moreno 1, 8
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-06-04 00:00:00 , DOI: 10.1021/acs.jctc.8b00150 Hugo A. L. Filipe 1, 2 , Matti Javanainen 3, 4 , Armindo Salvador 1, 2 , Adelino M. Galvão 5 , Ilpo Vattulainen 3, 4, 6 , Luís M. S. Loura 1, 7 , Maria João Moreno 1, 8
Affiliation
|
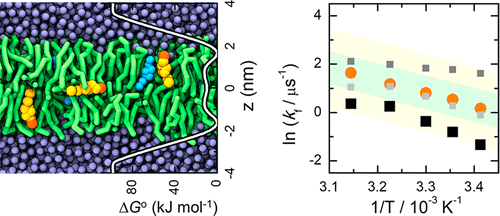
Accurately calculating rate constants of macroscopic chemical processes from molecular dynamics simulations is a long-sought but elusive goal. The problem is particularly relevant for processes occurring in biological systems, as is the case for ligand–protein and ligand–membrane interactions. Several formalisms to determine rate constants from easily accessible free-energy profiles [ΔGo(z)] of a molecule along a coordinate of interest have been proposed. However, their applicability for molecular interactions in condensed media has not been critically evaluated or validated. This work presents such evaluation and validation and introduces improved methodology. As a case study, we have characterized quantitatively the rate of translocation of cholesterol across 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayers. Translocation across lipid bilayers is the rate-limiting step in the permeation of most drugs through biomembranes. We use coarse-grained molecular dynamics simulations and different kinetic formalisms to calculate this rate constant. A self-consistent test of the applicability of various available formalisms is provided by comparing their predictions with the translocation rates obtained from actual events observed in long unrestrained simulations. To this effect, a novel procedure was used to obtain the effective rate constant, based on an analysis of time intervals between transitions among different states along the reaction coordinate. While most tested formalisms lead to results in reasonable agreement (within a factor of 5) with this effective rate constant, the most adequate one is based on the explicit relaxation frequencies from the transition state in the forward and backward directions along the reaction coordinate.
中文翻译:

从分子动力学模拟获得速率常数的方法的定量评估-胆固醇跨脂质双层的转运
从分子动力学模拟准确地计算宏观化学过程的速率常数是一个长期的但难以捉摸的目标。该问题与生物系统中发生的过程特别相关,配体-蛋白质和配体-膜的相互作用也是如此。从容易获得的自由能曲线[ ΔG o(z)]提出了一种沿着感兴趣坐标的分子。但是,它们在冷凝介质中分子相互作用的适用性尚未得到严格的评估或验证。这项工作提出了这种评估和确认,并介绍了改进的方法。作为案例研究,我们定量表征了1-palmitoyl-2-oleoyl- sn中胆固醇的转运速率-甘油-3-磷酸胆碱双层。跨脂质双层的转运是大多数药物通过生物膜渗透的速率限制步骤。我们使用粗粒度的分子动力学模拟和不同的动力学形式来计算该速率常数。通过比较它们的预测与从长期不受约束的模拟中观察到的实际事件获得的易位率,可以对各种可用形式主义的适用性进行自洽测试。为此,基于对沿着反应坐标的不同状态之间的跃迁之间的时间间隔的分析,使用了一种新颖的程序来获得有效速率常数。尽管大多数经过测试的形式主义都导致在此有效费率常数的基础上达成合理的一致(在5倍以内),
更新日期:2018-06-04
中文翻译:
从分子动力学模拟获得速率常数的方法的定量评估-胆固醇跨脂质双层的转运
从分子动力学模拟准确地计算宏观化学过程的速率常数是一个长期的但难以捉摸的目标。该问题与生物系统中发生的过程特别相关,配体-蛋白质和配体-膜的相互作用也是如此。从容易获得的自由能曲线[ ΔG o(z)]提出了一种沿着感兴趣坐标的分子。但是,它们在冷凝介质中分子相互作用的适用性尚未得到严格的评估或验证。这项工作提出了这种评估和确认,并介绍了改进的方法。作为案例研究,我们定量表征了1-palmitoyl-2-oleoyl- sn中胆固醇的转运速率-甘油-3-磷酸胆碱双层。跨脂质双层的转运是大多数药物通过生物膜渗透的速率限制步骤。我们使用粗粒度的分子动力学模拟和不同的动力学形式来计算该速率常数。通过比较它们的预测与从长期不受约束的模拟中观察到的实际事件获得的易位率,可以对各种可用形式主义的适用性进行自洽测试。为此,基于对沿着反应坐标的不同状态之间的跃迁之间的时间间隔的分析,使用了一种新颖的程序来获得有效速率常数。尽管大多数经过测试的形式主义都导致在此有效费率常数的基础上达成合理的一致(在5倍以内),










































 京公网安备 11010802027423号
京公网安备 11010802027423号