当前位置:
X-MOL 学术
›
Catal. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prediction of morphological changes of catalyst materials under reaction conditions by combined ab initio thermodynamics and microkinetic modelling†
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2018-06-01 00:00:00 , DOI: 10.1039/c8cy00583d Raffaele Cheula 1 , Aloysius Soon 2 , Matteo Maestri 1
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2018-06-01 00:00:00 , DOI: 10.1039/c8cy00583d Raffaele Cheula 1 , Aloysius Soon 2 , Matteo Maestri 1
Affiliation
|
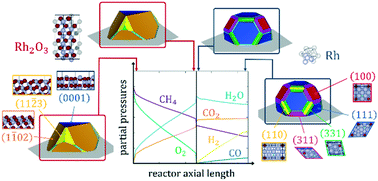
In this article, we couple microkinetic modelling, ab initio thermodynamics and Wulff–Kaishew construction to describe the structural variation of catalyst materials as a function of the chemical potential in the reactor. We focus specifically on experiments of catalytic partial oxidation (CPO) of methane on Rh/α-Al2O3. We employ a detailed structureless microkinetic model to calculate the profiles of the gaseous species molar fractions along the reactor coordinate and to select the most abundant reaction intermediates (MARIs) populating the catalyst surfaces in different zones of the reactor. Then, we calculate the most stable bulk and surface structures of the catalyst under different conditions of the reaction environment with density functional theory (DFT) calculations and ab initio thermodynamics, considering the presence of the MARIs on the catalyst surface in thermodynamic equilibrium with the partial pressures of their reservoirs in the gas phase surrounding the catalyst. Finally, we exploit the Wulff–Kaishew construction method to estimate the three-dimensional shape of the catalyst nanoparticles and the distribution of the active sites along the reactor coordinate. We find that the catalyst drastically modifies its morphology during CPO reaction by undergoing phase transition, in agreement with spectroscopy studies reported in the literature. The framework is also successfully applied for the analysis and interpretation of chemisorption experiments for catalyst characterization. These results demonstrate the crucial importance of rigorously accounting for the structural effect in microkinetic modeling simulations and pave the way towards the development of structure-dependent microkinetic analysis of catalytic processes.
中文翻译:

通过组合从头热力学和微动力学建模预测反应条件下催化剂材料的形态变化†
在本文中,我们结合微动力学建模、从头算热力学和 Wulff-Kaishew 结构来描述催化剂材料的结构变化作为反应器中化学势的函数。我们特别关注甲烷在 Rh/α-Al 2 O 3上的催化部分氧化(CPO)实验。我们采用详细的无结构微动力学模型来计算沿反应器坐标的气态物质摩尔分数的分布,并选择填充反应器不同区域的催化剂表面的最丰富的反应中间体(MARI)。然后,我们通过密度泛函理论(DFT)计算和从头算热力学计算了在不同反应环境条件下催化剂最稳定的体积和表面结构,考虑到催化剂表面上MARIs的存在与部分热力学平衡催化剂周围气相中储层的压力。最后,我们利用 Wulff-Kaishew 构建方法来估计催化剂纳米颗粒的三维形状以及活性位点沿反应器坐标的分布。我们发现催化剂在 CPO 反应过程中通过相变极大地改变了其形态,这与文献中报道的光谱研究一致。该框架还成功应用于催化剂表征化学吸附实验的分析和解释。这些结果证明了在微动力学建模模拟中严格考虑结构效应的至关重要性,并为催化过程的结构依赖性微动力学分析的发展铺平了道路。
更新日期:2018-06-01
中文翻译:
通过组合从头热力学和微动力学建模预测反应条件下催化剂材料的形态变化†
在本文中,我们结合微动力学建模、从头算热力学和 Wulff-Kaishew 结构来描述催化剂材料的结构变化作为反应器中化学势的函数。我们特别关注甲烷在 Rh/α-Al 2 O 3上的催化部分氧化(CPO)实验。我们采用详细的无结构微动力学模型来计算沿反应器坐标的气态物质摩尔分数的分布,并选择填充反应器不同区域的催化剂表面的最丰富的反应中间体(MARI)。然后,我们通过密度泛函理论(DFT)计算和从头算热力学计算了在不同反应环境条件下催化剂最稳定的体积和表面结构,考虑到催化剂表面上MARIs的存在与部分热力学平衡催化剂周围气相中储层的压力。最后,我们利用 Wulff-Kaishew 构建方法来估计催化剂纳米颗粒的三维形状以及活性位点沿反应器坐标的分布。我们发现催化剂在 CPO 反应过程中通过相变极大地改变了其形态,这与文献中报道的光谱研究一致。该框架还成功应用于催化剂表征化学吸附实验的分析和解释。这些结果证明了在微动力学建模模拟中严格考虑结构效应的至关重要性,并为催化过程的结构依赖性微动力学分析的发展铺平了道路。










































 京公网安备 11010802027423号
京公网安备 11010802027423号