当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Nonlocal Electron–Phonon Coupling in Prototypical Molecular Semiconductors from First Principles
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-05-31 00:00:00 , DOI: 10.1021/acs.jctc.8b00235 Xiaoyu Xie 1, 2 , Alejandro Santana-Bonilla 1 , Alessandro Troisi 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-05-31 00:00:00 , DOI: 10.1021/acs.jctc.8b00235 Xiaoyu Xie 1, 2 , Alejandro Santana-Bonilla 1 , Alessandro Troisi 1
Affiliation
|
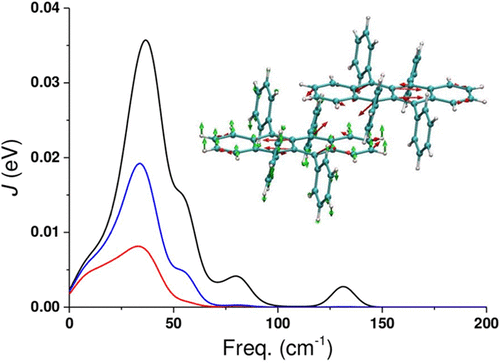
A substantial amount of evidence indicates a relevant role played by the nonlocal electron–phonon couplings in the mechanism of charge transport in organic semiconductors. In this work, we compute the nonlocal electron–phonon coupling for the prototypical molecular semiconductors rubrene and tetracene using the phonon modes obtained from ab initio methods. We do not make the rigid molecular approximation allowing a mixing of intra- and intermolecular modes, and we use a supercell approach to sample the momentum space. Indeed, we find that some low-frequency intramolecular modes are mixed with the rigid-molecule translations and rotations in the modes with the strongest electron–phonon coupling. To rationalize the results we propose a convenient decomposition of the delocalized lattice modes into molecular-based modes.
中文翻译:

基于第一性原理的典型分子半导体中的非局域电子-声子耦合
大量证据表明,非局部电子-声子耦合在有机半导体中的电荷传输机制中起着相关作用。在这项工作中,我们使用从头算得到的声子模式来计算原型分子半导体红荧烯和并四苯的非局部电子-声子耦合。方法。我们不进行允许分子内和分子间模式混合的刚性分子逼近,而是使用超单元方法对动量空间进行采样。确实,我们发现一些低频分子内模式与刚性分子的平移和旋转混合在一起,具有最强的电子-声子耦合。为了使结果合理化,我们建议将离域晶格模式分解为基于分子的模式。
更新日期:2018-05-31
中文翻译:
基于第一性原理的典型分子半导体中的非局域电子-声子耦合
大量证据表明,非局部电子-声子耦合在有机半导体中的电荷传输机制中起着相关作用。在这项工作中,我们使用从头算得到的声子模式来计算原型分子半导体红荧烯和并四苯的非局部电子-声子耦合。方法。我们不进行允许分子内和分子间模式混合的刚性分子逼近,而是使用超单元方法对动量空间进行采样。确实,我们发现一些低频分子内模式与刚性分子的平移和旋转混合在一起,具有最强的电子-声子耦合。为了使结果合理化,我们建议将离域晶格模式分解为基于分子的模式。










































 京公网安备 11010802027423号
京公网安备 11010802027423号