当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Consistent Prediction of Mutation Effect on Drug Binding in HIV-1 Protease Using Alchemical Calculations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-05-30 00:00:00 , DOI: 10.1021/acs.jctc.7b01109 Tomas Bastys 1, 2 , Vytautas Gapsys 3 , Nadezhda T. Doncheva 1, 4 , Rolf Kaiser 5 , Bert L. de Groot 3 , Olga V. Kalinina 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-05-30 00:00:00 , DOI: 10.1021/acs.jctc.7b01109 Tomas Bastys 1, 2 , Vytautas Gapsys 3 , Nadezhda T. Doncheva 1, 4 , Rolf Kaiser 5 , Bert L. de Groot 3 , Olga V. Kalinina 1
Affiliation
|
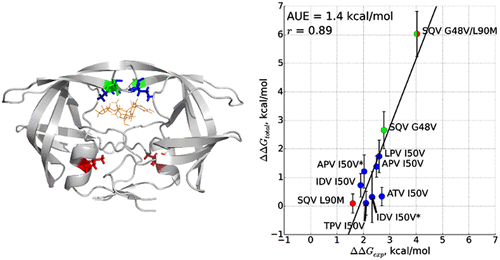
Despite a large number of antiretroviral drugs targeting HIV-1 protease for inhibition, mutations in this protein during the course of patient treatment can render them inefficient. This emerging resistance inspired numerous computational studies of the HIV-1 protease aimed at predicting the effect of mutations on drug binding in terms of free binding energy ΔG, as well as in mechanistic terms. In this study, we analyze ten different protease-inhibitor complexes carrying major resistance-associated mutations (RAMs) G48V, I50V, and L90M using molecular dynamics simulations. We demonstrate that alchemical free energy calculations can consistently predict the effect of mutations on drug binding. By explicitly probing different protonation states of the catalytic aspartic dyad, we reveal the importance of the correct choice of protonation state for the accuracy of the result. We also provide insight into how different mutations affect drug binding in their specific ways, with the unifying theme of how all of them affect the crucial drug binding regions of the protease.
中文翻译:

使用化学计算一致预测HIV-1蛋白酶对药物结合的突变作用
尽管有大量针对HIV-1蛋白酶的抗逆转录病毒药物具有抑制作用,但在患者治疗过程中该蛋白质的突变会使它们效率低下。这种新出现的抗药性激发了许多HIV-1蛋白酶的计算研究,旨在根据自由结合能ΔG预测突变对药物结合的影响。,以及机械术语。在这项研究中,我们使用分子动力学模拟分析了十种不同的蛋白酶抑制剂复合物,它们携带主要的抗药性相关突变(RAM)G48V,I50V和L90M。我们证明,炼金术的自由能计算可以一致地预测突变对药物结合的影响。通过显式探测催化天冬氨酸二元组的不同质子化状态,我们揭示了正确选择质子化状态对于结果准确性的重要性。我们还提供了有关不同突变如何以其特定方式影响药物结合的见解,并以它们全部如何影响蛋白酶的关键药物结合区为统一主题。
更新日期:2018-05-30
中文翻译:
使用化学计算一致预测HIV-1蛋白酶对药物结合的突变作用
尽管有大量针对HIV-1蛋白酶的抗逆转录病毒药物具有抑制作用,但在患者治疗过程中该蛋白质的突变会使它们效率低下。这种新出现的抗药性激发了许多HIV-1蛋白酶的计算研究,旨在根据自由结合能ΔG预测突变对药物结合的影响。,以及机械术语。在这项研究中,我们使用分子动力学模拟分析了十种不同的蛋白酶抑制剂复合物,它们携带主要的抗药性相关突变(RAM)G48V,I50V和L90M。我们证明,炼金术的自由能计算可以一致地预测突变对药物结合的影响。通过显式探测催化天冬氨酸二元组的不同质子化状态,我们揭示了正确选择质子化状态对于结果准确性的重要性。我们还提供了有关不同突变如何以其特定方式影响药物结合的见解,并以它们全部如何影响蛋白酶的关键药物结合区为统一主题。










































 京公网安备 11010802027423号
京公网安备 11010802027423号