当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electronic and Vibrational Spectroscopy of CsS
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-05-29 00:00:00 , DOI: 10.1021/acs.jpca.8b02543 Karim Elhadj Merabti 1 , Sihem Azizi 1 , Roberto Linguerri 2 , Gilberte Chambaud 2 , Muneerah Mogren Al-Mogren 3 , Majdi Hochlaf 2
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-05-29 00:00:00 , DOI: 10.1021/acs.jpca.8b02543 Karim Elhadj Merabti 1 , Sihem Azizi 1 , Roberto Linguerri 2 , Gilberte Chambaud 2 , Muneerah Mogren Al-Mogren 3 , Majdi Hochlaf 2
Affiliation
|
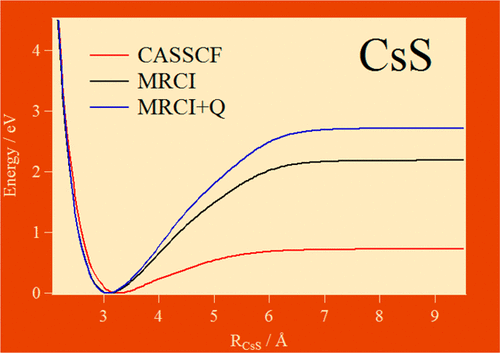
Using multi configurational ab initio methodologies, we compute the potential energy curves (PECs) of the lowest electronic states of the diatomic CsS. These computations are performed using internally contracted multireference interaction configuration including Davidson correction (MRCI+Q) with and without considering spin–orbit effects. The shapes of the PECs are governed by the interactions between the two ionic states, 2Σ+ and 2Π, correlating at large internuclear separations (RCsS) to the first ionic dissociation limit and the other electronic states correlating to the three lowest neutral dissociation limits. Computations show the importance of considering a large amount of electron correlation for the accurate description of the PECs and spectroscopy of this molecular system. As expected, these PECs are also strongly affected by the spin–orbit interaction. For the bound states, we report a set of spectroscopic parameters including equilibrium distances, dissociation energies, and vibrational and rotational constants. The effects of spin–orbit-induced changes on these parameters are also discussed. Moreover, we show that the 22Π state presents a “bowl” potential with a rather flat region extending to large RCsS distances. After being promoted to this state, wavepackets should undergo strong oscillations, similar to those observed by Zewail and co-workers for the NaI molecule. These should provide information on the shape of the PEC for the 22Π state and also on the couplings between this and the neighboring states.
中文翻译:

CsS的电子和振动光谱
使用多种配置的从头算方法,我们计算了双原子CsS的最低电子态的势能曲线(PEC)。这些计算是使用内部收缩的多参考相互作用配置(包括戴维森校正(MRCI + Q))进行的,并考虑或不考虑自旋轨道效应。胸肌的形状由相互作用两种离子状态,之间管辖2 Σ +和2 Π,在大核间分离相关(ř CSS)至第一个离子解离极限和与三个最低中性解离极限相关的其他电子态。计算表明,为了准确描述PEC和该分子系统的光谱学,必须考虑大量电子相关性。不出所料,这些PEC也受到自旋轨道相互作用的强烈影响。对于束缚态,我们报告了一组光谱参数,包括平衡距离,离解能以及振动和旋转常数。还讨论了自旋轨道引起的变化对这些参数的影响。此外,我们表明2 2Π状态呈现“碗状”电位,其相当平坦的区域扩展到较大的R CsS距离。在提升到这种状态后,波包应该经历强烈的振荡,这与Zewail及其同事对NaI分子所观察到的类似。这些应提供有关2 2Π状态的PEC形状以及此状态与相邻状态之间的耦合的信息。
更新日期:2018-05-29
中文翻译:
CsS的电子和振动光谱
使用多种配置的从头算方法,我们计算了双原子CsS的最低电子态的势能曲线(PEC)。这些计算是使用内部收缩的多参考相互作用配置(包括戴维森校正(MRCI + Q))进行的,并考虑或不考虑自旋轨道效应。胸肌的形状由相互作用两种离子状态,之间管辖2 Σ +和2 Π,在大核间分离相关(ř CSS)至第一个离子解离极限和与三个最低中性解离极限相关的其他电子态。计算表明,为了准确描述PEC和该分子系统的光谱学,必须考虑大量电子相关性。不出所料,这些PEC也受到自旋轨道相互作用的强烈影响。对于束缚态,我们报告了一组光谱参数,包括平衡距离,离解能以及振动和旋转常数。还讨论了自旋轨道引起的变化对这些参数的影响。此外,我们表明2 2Π状态呈现“碗状”电位,其相当平坦的区域扩展到较大的R CsS距离。在提升到这种状态后,波包应该经历强烈的振荡,这与Zewail及其同事对NaI分子所观察到的类似。这些应提供有关2 2Π状态的PEC形状以及此状态与相邻状态之间的耦合的信息。








































 京公网安备 11010802027423号
京公网安备 11010802027423号