JAMA Surgery ( IF 15.7 ) Pub Date : 2018-09-01 , DOI: 10.1001/jamasurg.2018.1153 Kepal N Patel 1 , Trevor E Angell 2 , Joshua Babiarz 3 , Neil M Barth 4, 5 , Thomas Blevins 6 , Quan-Yang Duh 7 , Ronald A Ghossein 8 , R Mack Harrell 9, 10, 11 , Jing Huang 3 , Giulia C Kennedy 3 , Su Yeon Kim 3 , Richard T Kloos 4 , Virginia A LiVolsi 12 , Gregory W Randolph 13 , Peter M Sadow 14 , Michael H Shanik 15 , Julie A Sosa 16 , S Thomas Traweek 17 , P Sean Walsh 3 , Duncan Whitney 3 , Michael W Yeh 18 , Paul W Ladenson 19
|
Importance Use of next-generation sequencing of RNA and machine learning algorithms can classify the risk of malignancy in cytologically indeterminate thyroid nodules to limit unnecessary diagnostic surgery.
Objective To measure the performance of a genomic sequencing classifier for cytologically indeterminate thyroid nodules.
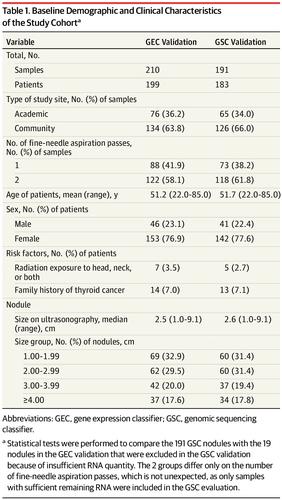
Design, Setting, and Participants A blinded validation study was conducted on a set of cytologically indeterminate thyroid nodules collected by fine-needle aspiration biopsy between June 2009 and December 2010 from 49 academic and community centers in the United States. All patients underwent surgery without genomic information and were assigned a histopathology diagnosis by an expert panel blinded to all genomic information. There were 210 potentially eligible thyroid biopsy samples with Bethesda III or IV indeterminate cytopathology that constituted a cohort previously used to validate the gene expression classifier. Of these, 191 samples (91.0%) had adequate residual RNA for validation of the genomic sequencing classifier. Algorithm development and independent validation occurred between August 2016 and May 2017.
Exposures Thyroid nodule surgical histopathology diagnosis by an expert panel blinded to all genomic data.
Main Outcomes and Measures The primary end point was measurement of genomic sequencing classifier sensitivity, specificity, and negative and positive predictive values in biopsies from Bethesda III and IV nodules. The secondary end point was measurement of classifier performance in biopsies from Bethesda II, V, and VI nodules.
Results Of the 183 included patients, 142 (77.6%) were women, and the mean (range) age was 51.7 (22.0-85.0) years. The genomic sequencing classifier had a sensitivity of 91% (95% CI, 79-98) and a specificity of 68% (95% CI, 60-76). At 24% cancer prevalence, the negative predictive value was 96% (95% CI, 90-99) and the positive predictive value was 47% (95% CI, 36-58).
Conclusions and Relevance The genomic sequencing classifier demonstrates high sensitivity and accuracy for identifying benign nodules. Its 36% increase in specificity compared with the gene expression classifier potentially increases the number of patients with benign nodules who can safely avoid unnecessary diagnostic surgery.
中文翻译:
基因组测序分类器在细胞学上不确定的甲状腺结节的术前诊断中的性能。
重要性 使用下一代RNA测序和机器学习算法可以对细胞学上不确定的甲状腺结节的恶性风险进行分类,以限制不必要的诊断性手术。
目的 评估基因组测序分类器在细胞学上不确定的甲状腺结节中的性能。
设计,设置和参与者 对2009年6月至2010年12月间从美国49个学术和社区中心通过细针穿刺活检收集的一组细胞学不确定的甲状腺结节进行了盲法验证研究。所有患者均接受了无基因组信息的手术,并由不了解所有基因组信息的专家小组进行了组织病理学诊断。有210例具有Bethesda III或IV不确定细胞病理学的潜在合格甲状腺活检样本,构成了先前用于验证基因表达分类器的队列。其中,有191个样品(占91.0%)具有足够的残留RNA,可用于验证基因组测序分类器。算法开发和独立验证发生在2016年8月至2017年5月之间。
暴露 甲状腺结节的手术组织病理学诊断由不了解所有基因组数据的专家小组诊断。
主要结果和措施 主要终点是测量Bethesda III和IV结节活检中的基因组测序分类器的灵敏度,特异性以及阴性和阳性预测值。次要终点是对Bethesda II,V和VI结节进行活检时分类器性能的测量。
结果 纳入的183例患者中,女性为142例(77.6%),平均(范围)年龄为51.7(22.0-85.0)岁。基因组测序分类器的灵敏度为91%(95%CI,79-98),特异性为68%(95%CI,60-76)。癌症患病率为24%时,阴性预测值为96%(95%CI,90-99),阳性预测值为47%(95%CI,36-58)。
结论和相关性 基因组测序分类器证明了对良性结节的高灵敏度和准确性。与基因表达分类器相比,其特异性提高了36%,这可能增加了可以安全避免不必要的诊断性手术的良性结节患者的数量。










































 京公网安备 11010802027423号
京公网安备 11010802027423号