当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Application of Semiempirical Methods to Transition Metal Complexes: Fast Results but Hard-to-Predict Accuracy
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-05-22 00:00:00 , DOI: 10.1021/acs.jctc.8b00018 Yury Minenkov 1, 2 , Dmitry I. Sharapa 3 , Luigi Cavallo 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-05-22 00:00:00 , DOI: 10.1021/acs.jctc.8b00018 Yury Minenkov 1, 2 , Dmitry I. Sharapa 3 , Luigi Cavallo 2
Affiliation
|
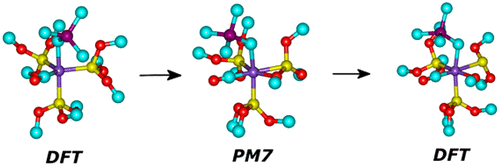
A series of semiempirical PM6* and PM7 methods has been tested in reproducing relative conformational energies of 27 realistic-size complexes of 16 different transition metals (TMs). An analysis of relative energies derived from single-point energy evaluations on density functional theory (DFT) optimized conformers revealed pronounced deviations between semiempirical and DFT methods, indicating a fundamental difference in potential energy surfaces (PES). To identify the origin of the deviation, we compared fully optimized PM7 and respective DFT conformers. For many complexes, differences in PM7 and DFT conformational energies have been confirmed often manifesting themselves in false coordination of some atoms (H, O) to TMs and chemical transformations/distortion of coordination center geometry in PM7 structures. Despite geometry optimization with fixed coordination center geometry leading to some improvements in conformational energies, the resulting accuracy is still too low to recommend explored semiempirical methods for out-of-the-box conformational search/sampling: careful testing is always needed.
中文翻译:

半经验方法在过渡金属配合物中的应用:快速的结果,但难以预测的准确性
在再现16种不同过渡金属(TM)的27种实际大小的络合物的相对构象能方面,已经测试了一系列的半经验PM6 *和PM7方法。根据密度泛函理论(DFT)优化构象从单点能量评估得出的相对能量分析显示,半经验方法和DFT方法之间存在明显的偏差,表明势能面(PES)存在根本差异。为了确定偏差的来源,我们比较了完全优化的PM7和相应的DFT构象异构体。对于许多络合物,已确认PM7和DFT构象能的差异通常表现为某些原子(H,O)与TM的错误配位以及PM7结构中配位中心几何形状的化学转化/变形。
更新日期:2018-05-22
中文翻译:
半经验方法在过渡金属配合物中的应用:快速的结果,但难以预测的准确性
在再现16种不同过渡金属(TM)的27种实际大小的络合物的相对构象能方面,已经测试了一系列的半经验PM6 *和PM7方法。根据密度泛函理论(DFT)优化构象从单点能量评估得出的相对能量分析显示,半经验方法和DFT方法之间存在明显的偏差,表明势能面(PES)存在根本差异。为了确定偏差的来源,我们比较了完全优化的PM7和相应的DFT构象异构体。对于许多络合物,已确认PM7和DFT构象能的差异通常表现为某些原子(H,O)与TM的错误配位以及PM7结构中配位中心几何形状的化学转化/变形。










































 京公网安备 11010802027423号
京公网安备 11010802027423号