当前位置:
X-MOL 学术
›
ACS Synth. Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
“Site and Mutation”-Specific Predictions Enable Minimal Directed Evolution Libraries
ACS Synthetic Biology ( IF 3.7 ) Pub Date : 2018-05-21 00:00:00 , DOI: 10.1021/acssynbio.7b00359 Jeffrey C Moore , Agustina Rodriguez-Granillo , Alejandro Crespo , Sridhar Govindarajan 1 , Mark Welch 1 , Kaori Hiraga , Katrina Lexa , Nicholas Marshall , Matthew D. Truppo
ACS Synthetic Biology ( IF 3.7 ) Pub Date : 2018-05-21 00:00:00 , DOI: 10.1021/acssynbio.7b00359 Jeffrey C Moore , Agustina Rodriguez-Granillo , Alejandro Crespo , Sridhar Govindarajan 1 , Mark Welch 1 , Kaori Hiraga , Katrina Lexa , Nicholas Marshall , Matthew D. Truppo
Affiliation
|
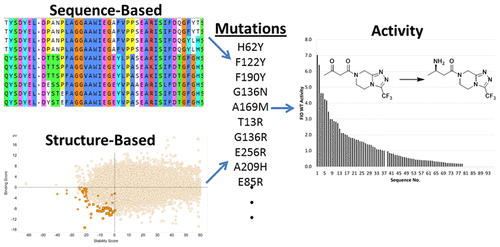
Directed evolution experiments designed to improve the activity of a biocatalyst have increased in sophistication from the early days of completely random mutagenesis. Sequence-based and structure-based methods have been developed to identify “hotspot” positions that when randomized provide a higher frequency of beneficial mutations that improve activity. These focused mutagenesis methods reduce library sizes and therefore reduce screening burden, accelerating the rate of finding improved enzymes. Looking for further acceleration in finding improved enzymes, we investigated whether two existing methods, one sequence-based (Protein GPS) and one structure-based (using Bioluminate and MOE), were sufficiently predictive to provide not just the hotspot position, but also the amino acid substitution that improved activity at that position. By limiting the libraries to variants that contained only specific amino acid substitutions, library sizes were kept to less than 100 variants. For an initial round of ATA-117 R-selective transaminase evolution, we found that the methods used produced libraries where 9% and 18% of the amino acid substitutions chosen were amino acids that improved reaction performance in lysates. The ability to create combinations of mutations as part of the initial design was confounded by the relatively large number of predicted mutations that were inactivating (30% and 45% for the sequence-based and structure-based methods, respectively). Despite this, combining several mutations identified within a given method produced variant lysates 7- and 9-fold more active than the wild-type lysate, highlighting the capability of mutations chosen this way to generate large advances in activity in addition to the reductions in screening.
中文翻译:

特定于“站点和突变”的预测可实现最少的定向进化库
从完全随机诱变的早期开始,旨在改善生物催化剂活性的定向进化实验就变得越来越复杂。已经开发了基于序列和基于结构的方法来识别“热点”位置,这些位置在随机化时可提供更高频率的有益突变,从而改善活性。这些集中的诱变方法减少了文库的大小,因此减少了筛选负担,加快了发现改良酶的速度。为了寻求进一步加速寻找改良酶的方法,我们调查了两种现有方法(一种基于序列的方法(蛋白质GPS)和一种基于结构的方法(使用Bioluminate和MOE))是否具有足够的预测能力,不仅可以提供热点位置,而且还可以提供氨基酸取代,可提高该位置的活性。通过将文库限制为仅包含特定氨基酸取代的变体,文库大小可保持小于100个变体。对于第一轮的ATA-117R选择性转氨酶的进化,我们发现所使用的方法产生的文库中,选择的9%和18%的氨基酸取代是改善了裂解物中反应性能的氨基酸。作为初始设计的一部分,创建突变组合的能力被大量失活的预测突变所混淆(基于序列的方法和基于结构的方法分别为30%和45%)。尽管如此,结合在给定方法中鉴定的几个突变产生的变异裂解物比野生型裂解物的活性高7到9倍,突出了以这种方式选择的突变的能力,除了降低了筛选效率外,还产生了较大的活性。
更新日期:2018-05-21
中文翻译:
特定于“站点和突变”的预测可实现最少的定向进化库
从完全随机诱变的早期开始,旨在改善生物催化剂活性的定向进化实验就变得越来越复杂。已经开发了基于序列和基于结构的方法来识别“热点”位置,这些位置在随机化时可提供更高频率的有益突变,从而改善活性。这些集中的诱变方法减少了文库的大小,因此减少了筛选负担,加快了发现改良酶的速度。为了寻求进一步加速寻找改良酶的方法,我们调查了两种现有方法(一种基于序列的方法(蛋白质GPS)和一种基于结构的方法(使用Bioluminate和MOE))是否具有足够的预测能力,不仅可以提供热点位置,而且还可以提供氨基酸取代,可提高该位置的活性。通过将文库限制为仅包含特定氨基酸取代的变体,文库大小可保持小于100个变体。对于第一轮的ATA-117R选择性转氨酶的进化,我们发现所使用的方法产生的文库中,选择的9%和18%的氨基酸取代是改善了裂解物中反应性能的氨基酸。作为初始设计的一部分,创建突变组合的能力被大量失活的预测突变所混淆(基于序列的方法和基于结构的方法分别为30%和45%)。尽管如此,结合在给定方法中鉴定的几个突变产生的变异裂解物比野生型裂解物的活性高7到9倍,突出了以这种方式选择的突变的能力,除了降低了筛选效率外,还产生了较大的活性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号