当前位置:
X-MOL 学术
›
ACS Infect. Dis.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanism of Action of Mycobacterium tuberculosis Gyrase Inhibitors: A Novel Class of Gyrase Poisons
ACS Infectious Diseases ( IF 4.0 ) Pub Date : 2018-05-10 00:00:00 , DOI: 10.1021/acsinfecdis.8b00035 Elizabeth G. Gibson , Tim R. Blower 1 , Monica Cacho 2 , Ben Bax 3 , James M. Berger 1 , Neil Osheroff 4
ACS Infectious Diseases ( IF 4.0 ) Pub Date : 2018-05-10 00:00:00 , DOI: 10.1021/acsinfecdis.8b00035 Elizabeth G. Gibson , Tim R. Blower 1 , Monica Cacho 2 , Ben Bax 3 , James M. Berger 1 , Neil Osheroff 4
Affiliation
|
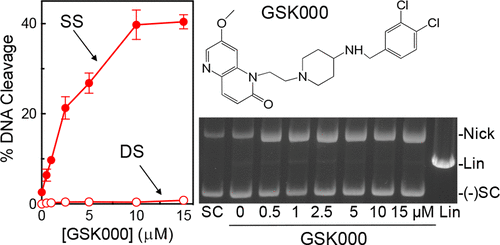
Tuberculosis is one of the leading causes of morbidity worldwide, and the incidences of drug resistance and intolerance are prevalent. Thus, there is a desperate need for the development of new antitubercular drugs. Mycobacterium tuberculosis gyrase inhibitors (MGIs) are napthyridone/aminopiperidine-based drugs that display activity against M. tuberculosis cells and tuberculosis in mouse models [Blanco, D., et al. (2015) Antimicrob. Agents Chemother. 59, 1868–1875]. Genetic and mutagenesis studies suggest that gyrase, which is the target for fluoroquinolone antibacterials, is also the target for MGIs. However, little is known regarding the interaction of these drugs with the bacterial type II enzyme. Therefore, we examined the effects of two MGIs, GSK000 and GSK325, on M. tuberculosis gyrase. MGIs greatly enhanced DNA cleavage mediated by the bacterial enzyme. In contrast to fluoroquinolones (which induce primarily double-stranded breaks), MGIs induced only single-stranded DNA breaks under a variety of conditions. MGIs work by stabilizing covalent gyrase-cleaved DNA complexes and appear to suppress the ability of the enzyme to induce double-stranded breaks. The drugs displayed little activity against type II topoisomerases from several other bacterial species, suggesting that these drugs display specificity for M. tuberculosis gyrase. Furthermore, MGIs maintained activity against M. tuberuclosis gyrase enzymes that contained the three most common fluoroquinolone resistance mutations seen in the clinic and displayed no activity against human topoisomerase IIα. These findings suggest that MGIs have potential as antitubercular drugs, especially in the case of fluoroquinolone-resistant disease.
中文翻译:

结核分枝杆菌促旋酶抑制剂的作用机制:一类新型的促旋酶毒物。
结核病是全世界发病率的主要原因之一,耐药性和不耐受性的发生率很高。因此,迫切需要开发新的抗结核药物。结核分枝杆菌促旋酶抑制剂(MGI)是基于萘啶酮/氨基哌啶的药物,在小鼠模型中显示出针对结核分枝杆菌细胞和结核的活性[Blanco,D.,et al。(2015)抗微生物剂。代理商Chemother。59(1868–1875年)。遗传和诱变研究表明,作为氟喹诺酮类抗菌药物靶标的促旋酶也是MGI的靶标。但是,关于这些药物与细菌II型酶的相互作用知之甚少。因此,我们检查了两种MGI(GSK000和GSK325)对结核分枝杆菌的影响旋转酶。MGI大大增强了由细菌酶介导的DNA切割。与氟喹诺酮类药物(主要诱导双链断裂)相反,MGI在多种条件下仅诱导单链DNA断裂。MGI通过稳定共价促旋酶切割的DNA复合物而起作用,并且似乎抑制了该酶诱导双链断裂的能力。这些药物对来自其他几种细菌的II型拓扑异构酶几乎没有活性,表明这些药物对结核分枝杆菌回旋酶显示出特异性。此外,MGI保持了抗结核分枝杆菌的活性在临床上见到的含有三种最常见的氟喹诺酮耐药性突变且不显示针对人拓扑异构酶IIα的活性的Gyrase酶。这些发现表明,MGI具有作为抗结核药物的潜力,特别是在氟喹诺酮耐药性疾病的情况下。
更新日期:2018-05-10
中文翻译:
结核分枝杆菌促旋酶抑制剂的作用机制:一类新型的促旋酶毒物。
结核病是全世界发病率的主要原因之一,耐药性和不耐受性的发生率很高。因此,迫切需要开发新的抗结核药物。结核分枝杆菌促旋酶抑制剂(MGI)是基于萘啶酮/氨基哌啶的药物,在小鼠模型中显示出针对结核分枝杆菌细胞和结核的活性[Blanco,D.,et al。(2015)抗微生物剂。代理商Chemother。59(1868–1875年)。遗传和诱变研究表明,作为氟喹诺酮类抗菌药物靶标的促旋酶也是MGI的靶标。但是,关于这些药物与细菌II型酶的相互作用知之甚少。因此,我们检查了两种MGI(GSK000和GSK325)对结核分枝杆菌的影响旋转酶。MGI大大增强了由细菌酶介导的DNA切割。与氟喹诺酮类药物(主要诱导双链断裂)相反,MGI在多种条件下仅诱导单链DNA断裂。MGI通过稳定共价促旋酶切割的DNA复合物而起作用,并且似乎抑制了该酶诱导双链断裂的能力。这些药物对来自其他几种细菌的II型拓扑异构酶几乎没有活性,表明这些药物对结核分枝杆菌回旋酶显示出特异性。此外,MGI保持了抗结核分枝杆菌的活性在临床上见到的含有三种最常见的氟喹诺酮耐药性突变且不显示针对人拓扑异构酶IIα的活性的Gyrase酶。这些发现表明,MGI具有作为抗结核药物的潜力,特别是在氟喹诺酮耐药性疾病的情况下。










































 京公网安备 11010802027423号
京公网安备 11010802027423号