当前位置:
X-MOL 学术
›
Acta Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Energetics of native defects, solute partitioning, and interfacial energy of Q precipitate in Al-Cu-Mg-Si alloys
Acta Materialia ( IF 8.3 ) Pub Date : 2018-08-01 , DOI: 10.1016/j.actamat.2018.05.031 Kyoungdoc Kim , Andrew Bobel , Vuk Brajuskovic , Bi-Cheng Zhou , Mike Walker , G.B. Olson , C. Wolverton
Acta Materialia ( IF 8.3 ) Pub Date : 2018-08-01 , DOI: 10.1016/j.actamat.2018.05.031 Kyoungdoc Kim , Andrew Bobel , Vuk Brajuskovic , Bi-Cheng Zhou , Mike Walker , G.B. Olson , C. Wolverton
|
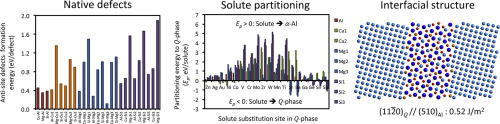
Abstract The compound Al3Cu2Mg9Si7, is known as the Q-phase and forms as a thermodynamically stable precipitate during aging in the quaternary Al-Cu-Mg-Si system. We perform atomic-scale density functional theory (DFT) calculations of defect properties, solute partitioning, and interfacial stability of the Al3Cu2Mg9Si7 (Q) precipitate. We find: (i) simple native point defect (i.e., vacancies and anti-sites) thermodynamics can partially explain the experimentally observed off-stoichiometry, such as the observed variation of compositions, Al3+δCu2Mg9-δSi7 (Mg-deficient and Al-rich) in experiment. (ii) Calculated solute-partitioning energies of common solutes allow us to define general rules for site-preference in the Q-phase in terms of electronic structure and atomic radius. To validate our DFT predictions, we perform atom-probe tomography (APT) experiments for six-different elements (Zn, Ni, Mn, Ti, V, and Zr). The results show that the partitioning behavior of solutes Ni, Zn, and Mn are consistent with DFT predictions, but the transition elements (Ti, V, and Zr), which are anomalously slow diffusers in Al, partition to the Q-phase in constrast to DFT partitioning energies. (iii) For the low energy interface (11 2 ¯ 0)Q//(510)Al observed in needle shaped Q-precipitate, we survey various terminations and orientations and derive a low-energy interfacial structure. We find this low-energy interfacial model has Cu atoms nearest to the interface, which is in agreement with previous literature on Cu interfacial segregation at the Q′//α-Al interface. The computed interfacial energy (0.52 J/m2) and the corresponding structure will be useful input to future multi-scale modeling of microstructural evolution.
中文翻译:

Al-Cu-Mg-Si合金中Q沉淀的本征缺陷能量、溶质分配和界面能
摘要 化合物 Al3Cu2Mg9Si7 被称为 Q 相,在四元 Al-Cu-Mg-Si 体系中时效期间形成热力学稳定的沉淀物。我们对 Al3Cu2Mg9Si7 (Q) 沉淀物的缺陷特性、溶质分配和界面稳定性进行原子级密度泛函理论 (DFT) 计算。我们发现:(i)简单的天然点缺陷(即空位和反位点)热力学可以部分解释实验观察到的非化学计量,例如观察到的成分变化,Al3+δCu2Mg9-δSi7(缺镁和铝-丰富)在实验中。(ii) 计算的常见溶质的溶质分配能允许我们根据电子结构和原子半径定义 Q 相中位点偏好的一般规则。为了验证我们的 DFT 预测,我们对六种不同的元素(Zn、Ni、Mn、Ti、V 和 Zr)进行原子探针断层扫描 (APT) 实验。结果表明,溶质 Ni、Zn 和 Mn 的分配行为与 DFT 预测一致,但过渡元素(Ti、V 和 Zr)是 Al 中异常缓慢的扩散体,相反则分配到 Q 相到 DFT 分配能量。(iii) 对于在针状 Q 沉淀物中观察到的低能量界面 (11 2 ¯ 0)Q//(510)Al,我们调查了各种终端和方向并推导出低能量界面结构。我们发现这种低能界面模型具有最靠近界面的 Cu 原子,这与先前关于 Q'//α-Al 界面处 Cu 界面偏析的文献一致。计算出的界面能 (0.
更新日期:2018-08-01
中文翻译:
Al-Cu-Mg-Si合金中Q沉淀的本征缺陷能量、溶质分配和界面能
摘要 化合物 Al3Cu2Mg9Si7 被称为 Q 相,在四元 Al-Cu-Mg-Si 体系中时效期间形成热力学稳定的沉淀物。我们对 Al3Cu2Mg9Si7 (Q) 沉淀物的缺陷特性、溶质分配和界面稳定性进行原子级密度泛函理论 (DFT) 计算。我们发现:(i)简单的天然点缺陷(即空位和反位点)热力学可以部分解释实验观察到的非化学计量,例如观察到的成分变化,Al3+δCu2Mg9-δSi7(缺镁和铝-丰富)在实验中。(ii) 计算的常见溶质的溶质分配能允许我们根据电子结构和原子半径定义 Q 相中位点偏好的一般规则。为了验证我们的 DFT 预测,我们对六种不同的元素(Zn、Ni、Mn、Ti、V 和 Zr)进行原子探针断层扫描 (APT) 实验。结果表明,溶质 Ni、Zn 和 Mn 的分配行为与 DFT 预测一致,但过渡元素(Ti、V 和 Zr)是 Al 中异常缓慢的扩散体,相反则分配到 Q 相到 DFT 分配能量。(iii) 对于在针状 Q 沉淀物中观察到的低能量界面 (11 2 ¯ 0)Q//(510)Al,我们调查了各种终端和方向并推导出低能量界面结构。我们发现这种低能界面模型具有最靠近界面的 Cu 原子,这与先前关于 Q'//α-Al 界面处 Cu 界面偏析的文献一致。计算出的界面能 (0.








































 京公网安备 11010802027423号
京公网安备 11010802027423号