当前位置:
X-MOL 学术
›
ChemMedChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Thermodynamic Insight into the Effects of Water Displacement and Rearrangement upon Ligand Modifications using Molecular Dynamics Simulations
ChemMedChem ( IF 3.6 ) Pub Date : 2018-05-30 , DOI: 10.1002/cmdc.201800093 Joel Wahl 1 , Martin Smieško 1
ChemMedChem ( IF 3.6 ) Pub Date : 2018-05-30 , DOI: 10.1002/cmdc.201800093 Joel Wahl 1 , Martin Smieško 1
Affiliation
|
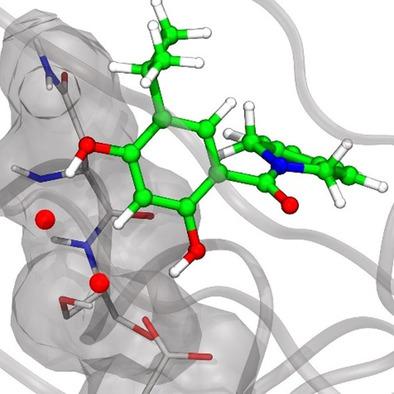
Computational methods, namely molecular dynamics (MD) simulations in combination with inhomogeneous fluid solvation theory (IFST) were used to retrospectively investigate various cases of ligand structure modifications that led to the displacement of binding site water molecules. Our findings are that water displacement per se is energetically unfavorable in the discussed examples, and that it is merely the fine balance between change in protein–ligand interaction energy, ligand solvation free energies, and binding site solvation free energies that determine if water displacement is favorable or not. We furthermore evaluated if we can reproduce experimental binding affinities by a computational approach combining changes in solvation free energies with changes in protein–ligand interaction energies and entropies. In two of the seven cases, this estimation led to large errors, implying that accurate predictions of relative binding free energies based on solvent thermodynamics is challenging. Nevertheless, MD simulations can provide insight regarding which water molecules can be targeted for displacement.
中文翻译:

使用分子动力学模拟对水置换和重排对配体修饰的影响的热力学见解
计算方法,即分子动力学(MD)模拟与非均匀流体溶剂化理论(IFST)相结合,用于回顾性研究导致配位点水分子置换的配体结构修饰的各种情况。我们的发现是在讨论的示例中水置换本身在能量上不利,并且仅仅是蛋白质-配体相互作用能的变化,配体溶剂化自由能和结合位点溶剂化自由能之间的良好平衡决定了水置换是否为有利与否。我们进一步评估了是否可以通过结合溶剂化自由能的变化与蛋白质-配体相互作用能和熵的变化的计算方法来重现实验的结合亲和力。在七个案例中的两个案例中,该估计导致较大的误差,这意味着基于溶剂热力学的相对结合自由能的准确预测具有挑战性。尽管如此,MD模拟仍可以提供有关哪些水分子可以作为置换目标的见解。
更新日期:2018-05-30
中文翻译:
使用分子动力学模拟对水置换和重排对配体修饰的影响的热力学见解
计算方法,即分子动力学(MD)模拟与非均匀流体溶剂化理论(IFST)相结合,用于回顾性研究导致配位点水分子置换的配体结构修饰的各种情况。我们的发现是在讨论的示例中水置换本身在能量上不利,并且仅仅是蛋白质-配体相互作用能的变化,配体溶剂化自由能和结合位点溶剂化自由能之间的良好平衡决定了水置换是否为有利与否。我们进一步评估了是否可以通过结合溶剂化自由能的变化与蛋白质-配体相互作用能和熵的变化的计算方法来重现实验的结合亲和力。在七个案例中的两个案例中,该估计导致较大的误差,这意味着基于溶剂热力学的相对结合自由能的准确预测具有挑战性。尽管如此,MD模拟仍可以提供有关哪些水分子可以作为置换目标的见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号