当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Tyrosine absorption spectroscopy: Backbone protonation effects on the side chain electronic properties
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-05-14 , DOI: 10.1002/jcc.25351 Sara Del Galdo 1 , Giordano Mancini 1, 2 , Isabella Daidone 3 , Laura Zanetti Polzi 3 , Andrea Amadei 4 , Vincenzo Barone 1, 2
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-05-14 , DOI: 10.1002/jcc.25351 Sara Del Galdo 1 , Giordano Mancini 1, 2 , Isabella Daidone 3 , Laura Zanetti Polzi 3 , Andrea Amadei 4 , Vincenzo Barone 1, 2
Affiliation
|
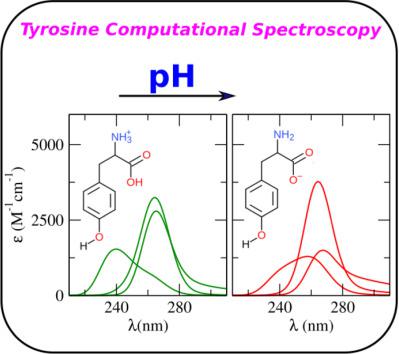
The UV–vis spectrum of Tyrosine and its response to different backbone protonation states have been studied by applying the Perturbed Matrix Method (PMM) in conjunction with molecular dynamics (MD) simulations. Herein, we theoretically reproduce the UV–vis absorption spectrum of aqueous solution of Tyrosine in its zwitterionic, anionic and cationic forms, as well as of aqua‐p‐Cresol (i.e., the moiety that constitutes the side chain portion of Tyrosine). To achieve a better accuracy in the MD sampling, the Tyrosine Force Field (FF) parameters were derived de novo via quantum mechanical calculations. The UV–vis absorption spectra are computed considering the occurring electronic transitions in the vertical approximation for each of the chromophore configurations sampled by the classical MD simulations, thus including the effects of the chromophore semiclassical structural fluctuations. Finally, the explicit treatment of the perturbing effect of the embedding environment permits to fully model the inhomogeneous bandwidth of the electronic spectra. Comparison between our theoretical–computational results and experimental data shows that the used model captures the essential features of the spectroscopic process, thus allowing to perform further analysis on the strict relationship between the quantum properties of the chromophore and the different embedding environments. © 2018 Wiley Periodicals, Inc.
中文翻译:

酪氨酸吸收光谱:主链质子化对侧链电子特性的影响
通过应用微扰矩阵法 (PMM) 结合分子动力学 (MD) 模拟,研究了酪氨酸的紫外可见光谱及其对不同骨架质子化状态的响应。在这里,我们理论上再现了两性离子、阴离子和阳离子形式的酪氨酸水溶液以及水对甲酚(即构成酪氨酸侧链部分的部分)的紫外可见吸收光谱。为了在 MD 采样中获得更好的精度,酪氨酸力场 (FF) 参数是通过量子力学计算从头推导出来的。考虑到经典 MD 模拟采样的每个发色团配置的垂直近似中发生的电子跃迁,计算 UV-vis 吸收光谱,因此包括发色团半经典结构波动的影响。最后,对嵌入环境的扰动效应的显式处理允许对电子光谱的非均匀带宽进行完全建模。我们的理论计算结果与实验数据之间的比较表明,所使用的模型捕获了光谱过程的基本特征,从而可以对发色团的量子特性与不同嵌入环境之间的严格关系进行进一步分析。© 2018 Wiley Periodicals, Inc. 我们的理论计算结果与实验数据之间的比较表明,所使用的模型捕获了光谱过程的基本特征,从而可以对发色团的量子特性与不同嵌入环境之间的严格关系进行进一步分析。© 2018 Wiley Periodicals, Inc. 我们的理论计算结果与实验数据之间的比较表明,所使用的模型捕获了光谱过程的基本特征,从而可以对发色团的量子特性与不同嵌入环境之间的严格关系进行进一步分析。© 2018 Wiley Periodicals, Inc.
更新日期:2018-05-14
中文翻译:
酪氨酸吸收光谱:主链质子化对侧链电子特性的影响
通过应用微扰矩阵法 (PMM) 结合分子动力学 (MD) 模拟,研究了酪氨酸的紫外可见光谱及其对不同骨架质子化状态的响应。在这里,我们理论上再现了两性离子、阴离子和阳离子形式的酪氨酸水溶液以及水对甲酚(即构成酪氨酸侧链部分的部分)的紫外可见吸收光谱。为了在 MD 采样中获得更好的精度,酪氨酸力场 (FF) 参数是通过量子力学计算从头推导出来的。考虑到经典 MD 模拟采样的每个发色团配置的垂直近似中发生的电子跃迁,计算 UV-vis 吸收光谱,因此包括发色团半经典结构波动的影响。最后,对嵌入环境的扰动效应的显式处理允许对电子光谱的非均匀带宽进行完全建模。我们的理论计算结果与实验数据之间的比较表明,所使用的模型捕获了光谱过程的基本特征,从而可以对发色团的量子特性与不同嵌入环境之间的严格关系进行进一步分析。© 2018 Wiley Periodicals, Inc. 我们的理论计算结果与实验数据之间的比较表明,所使用的模型捕获了光谱过程的基本特征,从而可以对发色团的量子特性与不同嵌入环境之间的严格关系进行进一步分析。© 2018 Wiley Periodicals, Inc. 我们的理论计算结果与实验数据之间的比较表明,所使用的模型捕获了光谱过程的基本特征,从而可以对发色团的量子特性与不同嵌入环境之间的严格关系进行进一步分析。© 2018 Wiley Periodicals, Inc.


























 京公网安备 11010802027423号
京公网安备 11010802027423号