当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Partition coefficients of methylated DNA bases obtained from free energy calculations with molecular electron density derived atomic charges
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-05-11 , DOI: 10.1002/jcc.25347 A. Lara 1 , M. Riquelme 1 , E. Vöhringer-Martinez 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-05-11 , DOI: 10.1002/jcc.25347 A. Lara 1 , M. Riquelme 1 , E. Vöhringer-Martinez 1
Affiliation
|
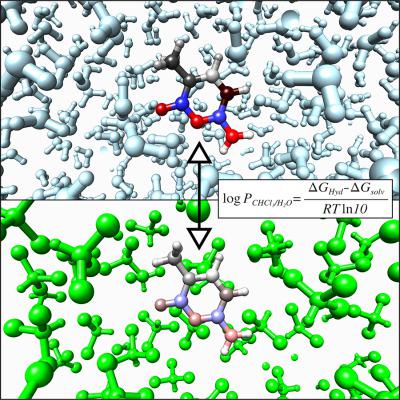
Partition coefficients serve in various areas as pharmacology and environmental sciences to predict the hydrophobicity of different substances. Recently, they have also been used to address the accuracy of force fields for various organic compounds and specifically the methylated DNA bases. In this study, atomic charges were derived by different partitioning methods (Hirshfeld and Minimal Basis Iterative Stockholder) directly from the electron density obtained by electronic structure calculations in a vacuum, with an implicit solvation model or with explicit solvation taking the dynamics of the solute and the solvent into account. To test the ability of these charges to describe electrostatic interactions in force fields for condensed phases, the original atomic charges of the AMBER99 force field were replaced with the new atomic charges and combined with different solvent models to obtain the hydration and chloroform solvation free energies by molecular dynamics simulations. Chloroform–water partition coefficients derived from the obtained free energies were compared to experimental and previously reported values obtained with the GAFF or the AMBER‐99 force field. The results show that good agreement with experimental data is obtained when the polarization of the electron density by the solvent has been taken into account, and when the energy needed to polarize the electron density of the solute has been considered in the transfer free energy. These results were further confirmed by hydration free energies of polar and aromatic amino acid side chain analogs. Comparison of the two partitioning methods, Hirshfeld‐I and Minimal Basis Iterative Stockholder (MBIS), revealed some deficiencies in the Hirshfeld‐I method related to the unstable isolated anionic nitrogen pro‐atom used in the method. Hydration free energies and partitioning coefficients obtained with atomic charges from the MBIS partitioning method accounting for polarization by the implicit solvation model are in good agreement with the experimental values. © 2018 Wiley Periodicals, Inc.
中文翻译:

从分子电子密度衍生的原子电荷计算自由能获得的甲基化 DNA 碱基的分配系数
分配系数在药理学和环境科学等各个领域都可以用于预测不同物质的疏水性。最近,它们还被用于解决各种有机化合物,特别是甲基化 DNA 碱基的力场的准确性。在这项研究中,原子电荷是通过不同的分配方法(Hirshfeld 和 Minimal Basis Iterative Stockholder)直接从在真空中通过电子结构计算获得的电子密度推导出来的,使用隐式溶剂化模型或显式溶剂化采用溶质和考虑到溶剂。为了测试这些电荷描述凝聚相力场中静电相互作用的能力,AMBER99 力场的原始原子电荷被替换为新的原子电荷,并结合不同的溶剂模型,通过分子动力学模拟获得水合和氯仿溶剂化自由能。将从获得的自由能得出的氯仿-水分配系数与用 GAFF 或 AMBER-99 力场获得的实验值和先前报告的值进行比较。结果表明,当考虑了溶剂对电子密度的极化时,以及在转移自由能中考虑了使溶质的电子密度极化所需的能量时,与实验数据吻合良好。极性和芳香族氨基酸侧链类似物的水合自由能进一步证实了这些结果。两种分配方法 Hirshfeld-I 和最小基迭代股东 (MBIS) 的比较揭示了 Hirshfeld-I 方法中与该方法中使用的不稳定的孤立阴离子氮原原子相关的一些缺陷。从 MBIS 分配方法获得的原子电荷的水合自由能和分配系数通过隐式溶剂化模型解释极化,与实验值非常一致。© 2018 Wiley Periodicals, Inc. 从 MBIS 分配方法获得的原子电荷的水合自由能和分配系数通过隐式溶剂化模型解释极化,与实验值非常一致。© 2018 Wiley Periodicals, Inc. 从 MBIS 分配方法获得的原子电荷的水合自由能和分配系数通过隐式溶剂化模型解释极化,与实验值非常一致。© 2018 Wiley Periodicals, Inc.
更新日期:2018-05-11
中文翻译:
从分子电子密度衍生的原子电荷计算自由能获得的甲基化 DNA 碱基的分配系数
分配系数在药理学和环境科学等各个领域都可以用于预测不同物质的疏水性。最近,它们还被用于解决各种有机化合物,特别是甲基化 DNA 碱基的力场的准确性。在这项研究中,原子电荷是通过不同的分配方法(Hirshfeld 和 Minimal Basis Iterative Stockholder)直接从在真空中通过电子结构计算获得的电子密度推导出来的,使用隐式溶剂化模型或显式溶剂化采用溶质和考虑到溶剂。为了测试这些电荷描述凝聚相力场中静电相互作用的能力,AMBER99 力场的原始原子电荷被替换为新的原子电荷,并结合不同的溶剂模型,通过分子动力学模拟获得水合和氯仿溶剂化自由能。将从获得的自由能得出的氯仿-水分配系数与用 GAFF 或 AMBER-99 力场获得的实验值和先前报告的值进行比较。结果表明,当考虑了溶剂对电子密度的极化时,以及在转移自由能中考虑了使溶质的电子密度极化所需的能量时,与实验数据吻合良好。极性和芳香族氨基酸侧链类似物的水合自由能进一步证实了这些结果。两种分配方法 Hirshfeld-I 和最小基迭代股东 (MBIS) 的比较揭示了 Hirshfeld-I 方法中与该方法中使用的不稳定的孤立阴离子氮原原子相关的一些缺陷。从 MBIS 分配方法获得的原子电荷的水合自由能和分配系数通过隐式溶剂化模型解释极化,与实验值非常一致。© 2018 Wiley Periodicals, Inc. 从 MBIS 分配方法获得的原子电荷的水合自由能和分配系数通过隐式溶剂化模型解释极化,与实验值非常一致。© 2018 Wiley Periodicals, Inc. 从 MBIS 分配方法获得的原子电荷的水合自由能和分配系数通过隐式溶剂化模型解释极化,与实验值非常一致。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号