当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Excited state dynamics and time-resolved photoelectron spectroscopy of para-xylylene
Faraday Discussions ( IF 3.3 ) Pub Date : 2018-05-11 , DOI: 10.1039/c8fd00083b Kevin Issler 1, 2, 3, 4 , Anja Röder 1, 2, 3, 4, 5 , Florian Hirsch 1, 2, 3, 4 , Lionel Poisson 5, 6, 7, 8, 9 , Ingo Fischer 1, 2, 3, 4 , Roland Mitrić 1, 2, 3, 4 , Jens Petersen 1, 2, 3, 4
Faraday Discussions ( IF 3.3 ) Pub Date : 2018-05-11 , DOI: 10.1039/c8fd00083b Kevin Issler 1, 2, 3, 4 , Anja Röder 1, 2, 3, 4, 5 , Florian Hirsch 1, 2, 3, 4 , Lionel Poisson 5, 6, 7, 8, 9 , Ingo Fischer 1, 2, 3, 4 , Roland Mitrić 1, 2, 3, 4 , Jens Petersen 1, 2, 3, 4
Affiliation
|
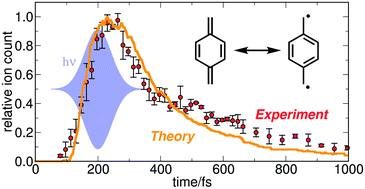
We investigated the excited-state dynamics of para-xylylene using a combination of field-induced surface hopping (FISH) simulations and time-resolved ionisation experiments. Our simulations predict an ultrafast decay of the initially excited bright state (S2/S3) to the S1 state on a sub-100 fs time scale, followed by return to the ground state within ∼1 ps. This is accompanied by a transient change of the biradical character of the molecule, as monitored by calculating natural orbital occupation numbers. Specifically, the initially low biradicality is increased by electronic excitation as well as by vibrational activation. Experimentally, para-xylylene was generated by pyrolysis from [2,2]paracyclophane and excited with 266 nm radiation into the S2/S3 bright state. The subsequent dynamics were followed using ionisation as the probe step, with both mass spectra and photoelectron spectra recorded as a function of pump–probe delay. The observed decay of photoelectron and photoion intensities closely matches the theoretical predictions and is consistent with the sequential mechanism found in the simulations. This mechanism exhibits characteristic signatures in both time-resolved mass and photoelectron spectra, in particular in the appearance of fragment ions that are exclusively generated from the S1 state. This allows for a separation of the S2 and S1 dynamics in the photoelectron and mass spectra. An excellent agreement between the observed and the simulated ion signal is observed.
中文翻译:

对二甲苯的激发态动力学和时间分辨光电子能谱
我们使用场致表面跳变(FISH)模拟和时间分辨电离实验的组合研究了对二甲苯的激发态动力学。我们的模拟预测在不到100 fs的时间尺度上,最初激发的亮态(S 2 / S 3)会迅速衰减到S 1状态,然后在约1 ps内返回到基态。这伴随着分子的双自由基特征的瞬时变化,如通过计算自然轨道占据数所监测的。具体地,通过电子激励以及通过振动激活来增加初始的低双自由基性。实验上,第-二甲苯是由[2,2]对环环烷经热解生成的,并在266 nm辐射下激发成S 2 / S 3亮态。随后的动力学过程是使用电离作为探测步骤,质谱和光电子谱均记录为泵浦-探针延迟的函数。观察到的光电子和光离子强度的衰减与理论预测值非常接近,并且与模拟中发现的顺序机制一致。该机理在时间分辨质谱和光电子谱中均表现出特征性特征,特别是在仅从S 1状态产生的碎片离子的外观中。这允许将S 2和S 1分开光电子和质谱的动力学。观察到的和模拟的离子信号之间有很好的一致性。
更新日期:2018-12-21
中文翻译:
对二甲苯的激发态动力学和时间分辨光电子能谱
我们使用场致表面跳变(FISH)模拟和时间分辨电离实验的组合研究了对二甲苯的激发态动力学。我们的模拟预测在不到100 fs的时间尺度上,最初激发的亮态(S 2 / S 3)会迅速衰减到S 1状态,然后在约1 ps内返回到基态。这伴随着分子的双自由基特征的瞬时变化,如通过计算自然轨道占据数所监测的。具体地,通过电子激励以及通过振动激活来增加初始的低双自由基性。实验上,第-二甲苯是由[2,2]对环环烷经热解生成的,并在266 nm辐射下激发成S 2 / S 3亮态。随后的动力学过程是使用电离作为探测步骤,质谱和光电子谱均记录为泵浦-探针延迟的函数。观察到的光电子和光离子强度的衰减与理论预测值非常接近,并且与模拟中发现的顺序机制一致。该机理在时间分辨质谱和光电子谱中均表现出特征性特征,特别是在仅从S 1状态产生的碎片离子的外观中。这允许将S 2和S 1分开光电子和质谱的动力学。观察到的和模拟的离子信号之间有很好的一致性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号