Drug Discovery Today ( IF 6.5 ) Pub Date : 2018-05-08 , DOI: 10.1016/j.drudis.2018.05.010 Yu-Chen Lo , Stefano E. Rensi , Wen Torng , Russ B. Altman
|
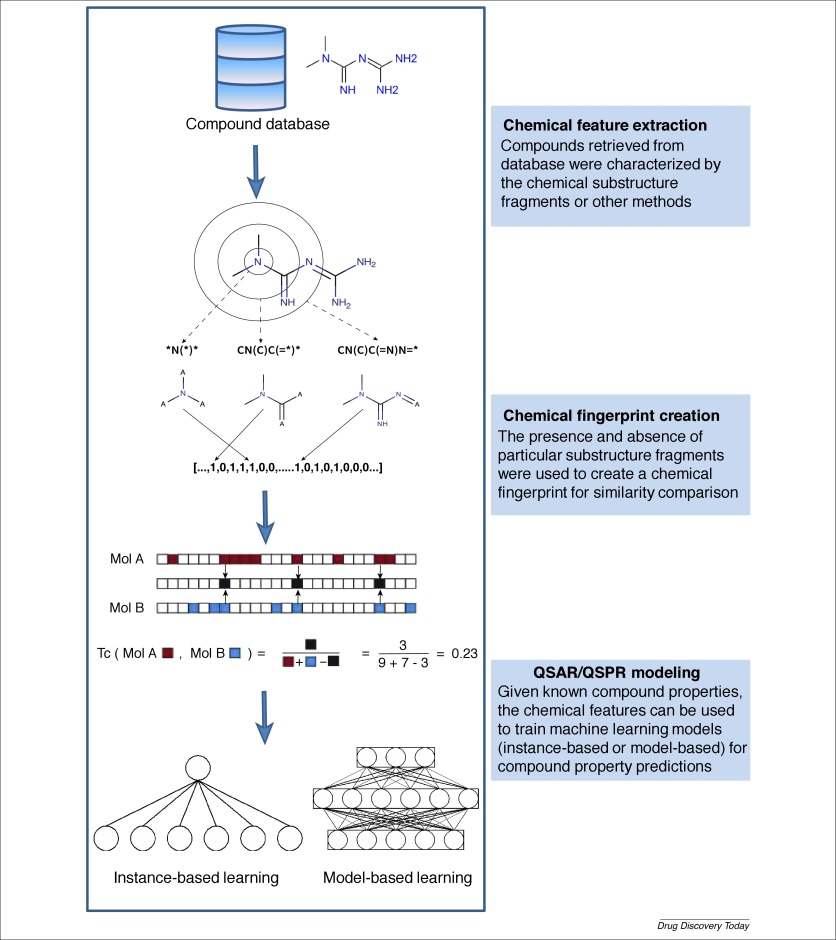
Chemoinformatics is an established discipline focusing on extracting, processing and extrapolating meaningful data from chemical structures. With the rapid explosion of chemical ‘big’ data from HTS and combinatorial synthesis, machine learning has become an indispensable tool for drug designers to mine chemical information from large compound databases to design drugs with important biological properties. To process the chemical data, we first reviewed multiple processing layers in the chemoinformatics pipeline followed by the introduction of commonly used machine learning models in drug discovery and QSAR analysis. Here, we present basic principles and recent case studies to demonstrate the utility of machine learning techniques in chemoinformatics analyses; and we discuss limitations and future directions to guide further development in this evolving field.
中文翻译:
化学信息学和药物发现中的机器学习
化学信息学是一门成熟的学科,致力于从化学结构中提取,处理和外推有意义的数据。随着来自HTS和组合合成的化学“大”数据的迅速爆炸,机器学习已成为药物设计人员从大型化合物数据库中挖掘化学信息以设计具有重要生物学特性的药物的必不可少的工具。为了处理化学数据,我们首先回顾了化学信息学管道中的多个处理层,然后在药物发现和QSAR分析中引入了常用的机器学习模型。在这里,我们介绍基本原理和最近的案例研究,以证明机器学习技术在化学信息学分析中的效用;










































 京公网安备 11010802027423号
京公网安备 11010802027423号