当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of Cyano Substitution on the Step-Edge Adsorption of Copper Phthalocyanine on Au(111)
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2018-05-04 00:00:00 , DOI: 10.1021/acs.jpcc.8b02178 Rejaul Sk , Srilatha Arra , Barun Dhara , Joel S. Miller 1 , Mukul Kabir , Aparna Deshpande
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2018-05-04 00:00:00 , DOI: 10.1021/acs.jpcc.8b02178 Rejaul Sk , Srilatha Arra , Barun Dhara , Joel S. Miller 1 , Mukul Kabir , Aparna Deshpande
Affiliation
|
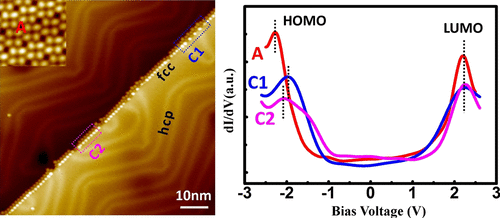
Step edges of single-crystal surfaces play an important role in tuning the electronic properties of the surfaces and in guiding the application of surfaces as catalytic reaction centers. Modification of step edges by molecular adsorption can be an effective strategy for bottom-up nanofabrication of surfaces. A detailed submolecular level understanding of step-edge adsorption is mandatory to exploit the properties of step edges for a variety of applications. Though a variety of phthalocyanine (Pc) molecules have been investigated on surfaces, there is a huge void in the literature about step-edge behavior of Pcs on surfaces. With this perspective, the adsorption characteristics of copper Pc (CuPc) and copperoctacyano Pc (CuPc(CN)8) have been investigated on Au(111) monoatomic (MA) step edges using low-temperature scanning tunneling microscopy (STM) and density functional theory (DFT) calculations. At very low coverage, the adsorption of CuPc and CuPc(CN)8 leads to the formation of one-dimensional chains along the step edge. At higher coverage, both CuPc and CuPc(CN)8 guided by tetramer unit cell formation self-assemble on flat terraces and cross over the step edge of Au(111). CuPc adsorption along MA step edge shows only one geometric configuration, whereas two different geometric configurations occur for CuPc(CN)8. The spectroscopic signature of these two configurations, probed using scanning tunneling spectroscopy (STS), manifests in a shift of the peak position of the highest occupied molecular orbital for the CuPc(CN)8 molecule at the MA step edge with respect to the molecule over the flat terrace of Au(111). The STM images simulated on the basis of DFT calculations for specific configurations agree with the experimental results. These findings also advance our understanding of the role played by the pendant groups of the Pc molecules in step-edge adsorption.
中文翻译:

氰基取代对酞菁铜在Au(111)上阶跃吸附的影响
单晶表面的台阶边缘在调节表面的电子性能和引导表面作为催化反应中心的应用中起着重要的作用。通过分子吸附修饰台阶边缘可能是表面自下而上纳米加工的有效策略。必须充分了解台阶边缘吸附的分子水平,才能在各种应用中利用台阶边缘的特性。尽管已对表面上的各种酞菁(Pc)分子进行了研究,但有关Pcs在表面上的台阶边缘行为的文献中仍存在巨大空白。从这个角度来看,铜Pc(CuPc)和八氰基铜Pc(CuPc(CN)8)已使用低温扫描隧道显微镜(STM)和密度泛函理论(DFT)计算在Au(111)单原子(MA)台阶边缘上进行了研究。在极低的覆盖率下,CuPc和CuPc(CN)8的吸附会导致沿台阶边缘形成一维链。在更高的覆盖率下,由四聚体晶胞形成引导的CuPc和CuPc(CN)8均在平坦平台上自组装并越过Au(111)的台阶边缘。沿着MA台阶边缘的CuPc吸附仅显示一种几何构型,而CuPc(CN)8发生两种不同的几何构型。使用扫描隧道光谱法(STS)探测到的这两种构型的光谱特征表明,相对于分子超过MA阶,CuPc(CN)8分子的最高占据分子轨道的峰位置在MA台阶边缘处发生了移动Au(111)的平坦露台。在特定配置下基于DFT计算得出的STM图像与实验结果吻合。这些发现也使我们进一步理解了Pc分子的侧基在台阶边缘吸附中的作用。
更新日期:2018-05-04
中文翻译:
氰基取代对酞菁铜在Au(111)上阶跃吸附的影响
单晶表面的台阶边缘在调节表面的电子性能和引导表面作为催化反应中心的应用中起着重要的作用。通过分子吸附修饰台阶边缘可能是表面自下而上纳米加工的有效策略。必须充分了解台阶边缘吸附的分子水平,才能在各种应用中利用台阶边缘的特性。尽管已对表面上的各种酞菁(Pc)分子进行了研究,但有关Pcs在表面上的台阶边缘行为的文献中仍存在巨大空白。从这个角度来看,铜Pc(CuPc)和八氰基铜Pc(CuPc(CN)8)已使用低温扫描隧道显微镜(STM)和密度泛函理论(DFT)计算在Au(111)单原子(MA)台阶边缘上进行了研究。在极低的覆盖率下,CuPc和CuPc(CN)8的吸附会导致沿台阶边缘形成一维链。在更高的覆盖率下,由四聚体晶胞形成引导的CuPc和CuPc(CN)8均在平坦平台上自组装并越过Au(111)的台阶边缘。沿着MA台阶边缘的CuPc吸附仅显示一种几何构型,而CuPc(CN)8发生两种不同的几何构型。使用扫描隧道光谱法(STS)探测到的这两种构型的光谱特征表明,相对于分子超过MA阶,CuPc(CN)8分子的最高占据分子轨道的峰位置在MA台阶边缘处发生了移动Au(111)的平坦露台。在特定配置下基于DFT计算得出的STM图像与实验结果吻合。这些发现也使我们进一步理解了Pc分子的侧基在台阶边缘吸附中的作用。








































 京公网安备 11010802027423号
京公网安备 11010802027423号