当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantum and classical IR spectra of (HCOOH)2, (DCOOH)2 and (DCOOD)2 using ab initio potential energy and dipole moment surfaces
Faraday Discussions ( IF 3.3 ) Pub Date : 2018-05-02 , DOI: 10.1039/c8fd00077h Chen Qu 1, 2, 3, 4, 5 , Joel M. Bowman 1, 2, 3, 4, 5
Faraday Discussions ( IF 3.3 ) Pub Date : 2018-05-02 , DOI: 10.1039/c8fd00077h Chen Qu 1, 2, 3, 4, 5 , Joel M. Bowman 1, 2, 3, 4, 5
Affiliation
|
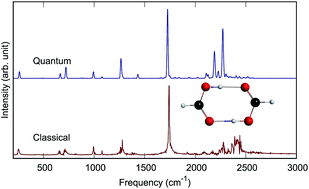
An accurate quantum mechanical description of the vibrational dynamics and IR spectra of molecules is illustrated here for the formic acid dimer, (HCOOH)2 and its isotopologues (DCOOH)2 and (DCOOD)2 in full dimensionality. The calculations make use of recent full-dimensional ab initio potential energy and dipole moment surfaces and are done with the code MULTIMODE. IR spectra are reported for the three dimers and also compared to available experimental spectra. In addition, standard classical and “semiclassically prepared” quasiclassical molecular dynamics calculations of the IR spectra of these complexes are reported and compared to the quantum spectra and also experiment. These comparisons indicate good accuracy of the MD spectra for sharp bands but not for the complex O–H stretch band, where the complex molecular dynamics band is upshifted from experiments by roughly 300 cm−1. For the fully deuterated dimer (DCOOD)2, the quantum spectral band for the O–D stretch sharpens relative to the O–H spectral bands in (HCOOH)2 and (DCOOH)2; however, the molecular dynamics OD stretch band does not exhibit this sharpening.
中文翻译:

量子和(HCOOH)的经典IR光谱2,(DCOOH)2和(DCOOD)2使用从头势能和偶极矩表面
此处以完整尺寸显示了甲酸二聚体(HCOOH)2及其同位素(DCOOH)2和(DCOOD)2的分子振动动力学和IR光谱的精确量子力学描述。计算使用了最近的全尺寸从头算势能和偶极矩表面,并使用代码MULTIMODE完成。报告了三个二聚体的红外光谱,并与可用的实验光谱进行了比较。此外,还报道了这些络合物的红外光谱的标准经典和“半经典制备”准经典分子动力学计算结果,并将其与量子光谱进行了比较,并进行了实验。这些比较表明,MD光谱对于尖峰带具有良好的准确性,但对于复杂的OH拉伸带却没有,因为复杂的分子动力学带比实验上移了约300 cm -1。对于完全氘化的二聚体(DCOOD)2,相对于(HCOOH)2和(DCOOH)中的O–H光谱带,O–D拉伸的量子光谱带变尖。2 ; 然而,分子动力学OD拉伸带没有表现出这种锐化。
更新日期:2018-12-21
中文翻译:
量子和(HCOOH)的经典IR光谱2,(DCOOH)2和(DCOOD)2使用从头势能和偶极矩表面
此处以完整尺寸显示了甲酸二聚体(HCOOH)2及其同位素(DCOOH)2和(DCOOD)2的分子振动动力学和IR光谱的精确量子力学描述。计算使用了最近的全尺寸从头算势能和偶极矩表面,并使用代码MULTIMODE完成。报告了三个二聚体的红外光谱,并与可用的实验光谱进行了比较。此外,还报道了这些络合物的红外光谱的标准经典和“半经典制备”准经典分子动力学计算结果,并将其与量子光谱进行了比较,并进行了实验。这些比较表明,MD光谱对于尖峰带具有良好的准确性,但对于复杂的OH拉伸带却没有,因为复杂的分子动力学带比实验上移了约300 cm -1。对于完全氘化的二聚体(DCOOD)2,相对于(HCOOH)2和(DCOOH)中的O–H光谱带,O–D拉伸的量子光谱带变尖。2 ; 然而,分子动力学OD拉伸带没有表现出这种锐化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号