当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Reliable and Performant Identification of Low-Energy Conformers in the Gas Phase and Water
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-05-02 00:00:00 , DOI: 10.1021/acs.jcim.8b00151 Anna Theresa Cavasin 1 , Alexander Hillisch 1 , Felix Uellendahl 1 , Sebastian Schneckener 2 , Andreas H. Göller 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-05-02 00:00:00 , DOI: 10.1021/acs.jcim.8b00151 Anna Theresa Cavasin 1 , Alexander Hillisch 1 , Felix Uellendahl 1 , Sebastian Schneckener 2 , Andreas H. Göller 1
Affiliation
|
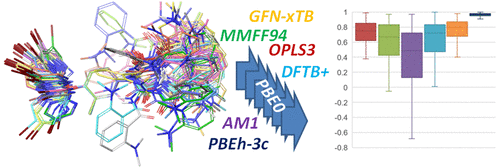
Prediction of compound properties from structure via quantitative structure–activity relationship and machine-learning approaches is an important computational chemistry task in small-molecule drug research. Though many such properties are dependent on three-dimensional structures or even conformer ensembles, the majority of models are based on descriptors derived from two-dimensional structures. Here we present results from a thorough benchmark study of force field, semiempirical, and density functional methods for the calculation of conformer energies in the gas phase and water solvation as a foundation for the correct identification of relevant low-energy conformers. We find that the tight-binding ansatz GFN-xTB shows the lowest error metrics and highest correlation to the benchmark PBE0-D3(BJ)/def2-TZVP in the gas phase for the computationally fast methods and that in solvent OPLS3 becomes comparable in performance. MMFF94, AM1, and DFTB+ perform worse, whereas the performance-optimized but far more expensive functional PBEh-3c yields energies almost perfectly correlated to the benchmark and should be used whenever affordable. On the basis of our findings, we have implemented a reliable and fast protocol for the identification of low-energy conformers of drug-like molecules in water that can be used for the quantification of strain energy and entropy contributions to target binding as well as for the derivation of conformer-ensemble-dependent molecular descriptors.
中文翻译:

气相和水中低能合格品的可靠且性能可靠的识别
通过定量结构-活性关系和机器学习方法从结构预测化合物性质是小分子药物研究中重要的计算化学任务。尽管许多此类属性都依赖于三维结构,甚至依赖于构象合体,但大多数模型都是基于从二维结构派生的描述符。在这里,我们从力场,半经验和密度泛函方法的彻底基准研究中获得结果,这些方法用于计算气相和水溶剂化中的构象能量,为正确识别相关的低能构象奠定了基础。我们发现紧密结合的ansatz GFN-xTB在气相中显示出最低的误差指标和与基准PBE0-D3(BJ)/ def2-TZVP的最高相关性,这对于快速计算方法而言是可比的,而在溶剂OPLS3中则表现出可比性。MMFF94,AM1和DFTB +的性能较差,而性能优化但价格昂贵得多的功能性PBEh-3c产生的能量几乎与基准完全相关,应在负担得起的情况下使用。根据我们的发现,我们已经实施了一种可靠,快速的方案,用于鉴定水中药物样分子的低能构象体,可用于量化应变能和熵对靶标结合的贡献,以及用于依赖于构象异构体集合的分子描述符的推导。
更新日期:2018-05-02
中文翻译:
气相和水中低能合格品的可靠且性能可靠的识别
通过定量结构-活性关系和机器学习方法从结构预测化合物性质是小分子药物研究中重要的计算化学任务。尽管许多此类属性都依赖于三维结构,甚至依赖于构象合体,但大多数模型都是基于从二维结构派生的描述符。在这里,我们从力场,半经验和密度泛函方法的彻底基准研究中获得结果,这些方法用于计算气相和水溶剂化中的构象能量,为正确识别相关的低能构象奠定了基础。我们发现紧密结合的ansatz GFN-xTB在气相中显示出最低的误差指标和与基准PBE0-D3(BJ)/ def2-TZVP的最高相关性,这对于快速计算方法而言是可比的,而在溶剂OPLS3中则表现出可比性。MMFF94,AM1和DFTB +的性能较差,而性能优化但价格昂贵得多的功能性PBEh-3c产生的能量几乎与基准完全相关,应在负担得起的情况下使用。根据我们的发现,我们已经实施了一种可靠,快速的方案,用于鉴定水中药物样分子的低能构象体,可用于量化应变能和熵对靶标结合的贡献,以及用于依赖于构象异构体集合的分子描述符的推导。










































 京公网安备 11010802027423号
京公网安备 11010802027423号