当前位置:
X-MOL 学术
›
ACS Chem. Neurosci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Proposing Novel MAO-B Hit Inhibitors Using Multidimensional Molecular Modeling Approaches and Application of Binary QSAR Models for Prediction of Their Therapeutic Activity, Pharmacokinetic and Toxicity Properties
ACS Chemical Neuroscience ( IF 4.1 ) Pub Date : 2018-04-19 00:00:00 , DOI: 10.1021/acschemneuro.8b00095 Yusuf Serhat Is 1, 2, 3 , Serdar Durdagi 1, 4 , Busecan Aksoydan 1, 4 , Mine Yurtsever 2
ACS Chemical Neuroscience ( IF 4.1 ) Pub Date : 2018-04-19 00:00:00 , DOI: 10.1021/acschemneuro.8b00095 Yusuf Serhat Is 1, 2, 3 , Serdar Durdagi 1, 4 , Busecan Aksoydan 1, 4 , Mine Yurtsever 2
Affiliation
|
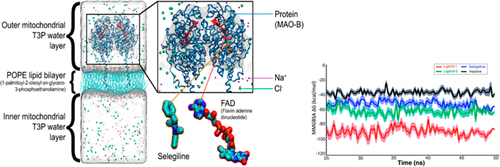
Monoamine oxidase (MAO) enzymes MAO-A and MAO-B play a critical role in the metabolism of monoamine neurotransmitters. Hence, MAO inhibitors are very important for the treatment of several neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS). In this study, 256 750 molecules from Otava Green Chemical Collection were virtually screened for their binding activities as MAO-B inhibitors. Two hit molecules were identified after applying different filters such as high docking scores and selectivity to MAO-B, desired pharmacokinetic profile predictions with binary quantitative structure–activity relationship (QSAR) models. Therapeutic activity prediction as well as pharmacokinetic and toxicity profiles were investigated using MetaCore/MetaDrug platform which is based on a manually curated database of molecular interactions, molecular pathways, gene–disease associations, chemical metabolism, and toxicity information. Particular therapeutic activity and toxic effect predictions are based on the ChemTree ability to correlate structural descriptors to that property using recursive partitioning algorithm. Molecular dynamics (MD) simulations were also performed to make more detailed assessments beyond docking studies. All these calculations were made not only to determine if studied molecules possess the potential to be a MAO-B inhibitor but also to find out whether they carry MAO-B selectivity versus MAO-A. The evaluation of docking results and pharmacokinetic profile predictions together with the MD simulations enabled us to identify one hit molecule (ligand 1, Otava ID: 3463218) which displayed higher selectivity toward MAO-B than a positive control selegiline which is a commercially used drug for PD therapeutic purposes.
中文翻译:

提出使用多维分子建模方法的新型MAO-B命中抑制剂,并应用二元QSAR模型预测其治疗活性,药代动力学和毒性
单胺氧化酶(MAO)酶MAO-A和MAO-B在单胺神经递质的代谢中起关键作用。因此,MAO抑制剂对于治疗几种神经退行性疾病(例如帕金森氏病(PD),阿尔茨海默氏病(AD)和肌萎缩性侧索硬化症(ALS))非常重要。在这项研究中,对Otava Green Chemical Collection的256750个分子作为MAO-B抑制剂的结合活性进行了虚拟筛选。在应用不同的过滤器(例如,高对接得分和对MAO-B的选择性),具有二元定量结构-活性关系(QSAR)模型的所需药代动力学概况预测后,鉴定出两个命中分子。使用MetaCore / MetaDrug平台研究了治疗活性预测以及药代动力学和毒性概况,该平台基于分子相互作用,分子途径,基因-疾病关联,化学代谢和毒性信息的手动管理数据库。特定的治疗活性和毒性效应预测是基于ChemTree使用递归分区算法将结构描述符与该特性相关联的能力。除对接研究外,还进行了分子动力学(MD)模拟以进行更详细的评估。所有这些计算不仅可以确定所研究的分子是否具有成为MAO-B抑制剂的潜力,还可以确定它们相对于MAO-A是否具有MAO-B选择性。参见图1,Otava ID:3462218),其对MAO-B表现出比作为商业上用于PD治疗目的的药物的阳性对照司来吉兰更高的选择性。
更新日期:2018-04-19
中文翻译:
提出使用多维分子建模方法的新型MAO-B命中抑制剂,并应用二元QSAR模型预测其治疗活性,药代动力学和毒性
单胺氧化酶(MAO)酶MAO-A和MAO-B在单胺神经递质的代谢中起关键作用。因此,MAO抑制剂对于治疗几种神经退行性疾病(例如帕金森氏病(PD),阿尔茨海默氏病(AD)和肌萎缩性侧索硬化症(ALS))非常重要。在这项研究中,对Otava Green Chemical Collection的256750个分子作为MAO-B抑制剂的结合活性进行了虚拟筛选。在应用不同的过滤器(例如,高对接得分和对MAO-B的选择性),具有二元定量结构-活性关系(QSAR)模型的所需药代动力学概况预测后,鉴定出两个命中分子。使用MetaCore / MetaDrug平台研究了治疗活性预测以及药代动力学和毒性概况,该平台基于分子相互作用,分子途径,基因-疾病关联,化学代谢和毒性信息的手动管理数据库。特定的治疗活性和毒性效应预测是基于ChemTree使用递归分区算法将结构描述符与该特性相关联的能力。除对接研究外,还进行了分子动力学(MD)模拟以进行更详细的评估。所有这些计算不仅可以确定所研究的分子是否具有成为MAO-B抑制剂的潜力,还可以确定它们相对于MAO-A是否具有MAO-B选择性。参见图1,Otava ID:3462218),其对MAO-B表现出比作为商业上用于PD治疗目的的药物的阳性对照司来吉兰更高的选择性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号