当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational and Spectroscopic Analysis of β-Indandione Modified Zinc Porphyrins
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-04-19 00:00:00 , DOI: 10.1021/acs.jpca.8b02746 Joseph I. Mapley 1 , Pawel Wagner 2 , David L. Officer 2 , Keith C. Gordon 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-04-19 00:00:00 , DOI: 10.1021/acs.jpca.8b02746 Joseph I. Mapley 1 , Pawel Wagner 2 , David L. Officer 2 , Keith C. Gordon 1
Affiliation
|
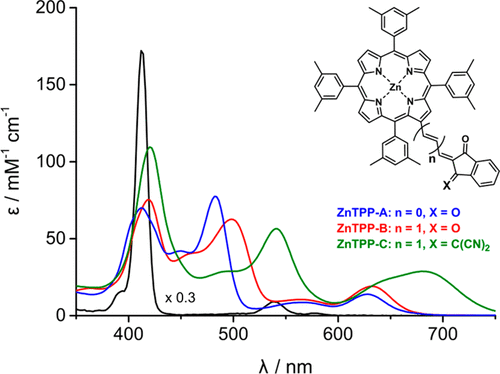
Porphyrins have characteristic optical properties which give them the potential to be used in a range of applications. In this study, a series of β-indandione modified zinc porphyrins, systematically changed in terms of linker length and substituent, resulted in absorption spectra that are dramatically different than that observed for the parent zinc porphyrin (ZnTXP, 5,10,15,20-tetrakis(3,5-dimethylphenyl)porphyrinato zinc(II)). These changes include strong absorptions at 420, 541, and 681 nm (110.2, 57.5, and 29.2 mM–1 cm–1, respectively) for the most perturbed compound. Computational studies were conducted and showed the different optical effects are due to a reorganization of molecular orbitals (MOs) away from Gouterman’s four-orbital model. The substituent effects alter both unoccupied and occupied MOs. An increased length of linker group raised the energy of the HOMO–2 such that it plays a significant role in the observed transitions. The degenerate LUMO (eg) set are split by substitution, and this splitting may be increased by use of a propylidenodinitrile group, which shows the lowest-energy transitions and the greatest spectral perturbation from the parent zinc porphyrin complex. These data are supported by resonance Raman spectroscopy studies which show distinct enhancement of phenyl modes for high-energy transitions and indandione modes for lower-energy transitions.
中文翻译:

β-茚满二酮修饰的锌卟啉的计算与光谱分析
卟啉具有独特的光学性质,使其有潜力在一系列应用中使用。在这项研究中,一系列的β-茚满二酮修饰的锌卟啉在接头长度和取代基方面发生了系统性的变化,从而导致吸收光谱与母体卟啉锌所观察到的显着不同(ZnTXP,5,10,15,20 -四(3,5-二甲基苯基)卟啉锌(II))。这些变化包括在420、541和681 nm(110.2、57.5和29.2 mM –1 cm –1,分别表示受干扰最大的化合物。进行了计算研究,结果表明不同的光学效应是由于分子结构偏离了Gouterman的四轨道模型而引起的。取代基效应同时改变了未占据的MO和被占据的MO。连接基团长度的增加增加了HOMO-2的能量,因此它在观察到的过渡中起着重要作用。简并LUMO(例如克集合通过取代分裂,并且该分裂可通过使用亚丙基去腈基团来增加,该基团显示出最低的能量跃迁和来自母卟啉锌配合物的最大光谱扰动。这些数据得到共振拉曼光谱研究的支持,共振拉曼光谱研究表明,对于高能跃迁,苯基模式显着增强;对于低能跃迁,茚满二酮模式显着增强。
更新日期:2018-04-19
中文翻译:
β-茚满二酮修饰的锌卟啉的计算与光谱分析
卟啉具有独特的光学性质,使其有潜力在一系列应用中使用。在这项研究中,一系列的β-茚满二酮修饰的锌卟啉在接头长度和取代基方面发生了系统性的变化,从而导致吸收光谱与母体卟啉锌所观察到的显着不同(ZnTXP,5,10,15,20 -四(3,5-二甲基苯基)卟啉锌(II))。这些变化包括在420、541和681 nm(110.2、57.5和29.2 mM –1 cm –1,分别表示受干扰最大的化合物。进行了计算研究,结果表明不同的光学效应是由于分子结构偏离了Gouterman的四轨道模型而引起的。取代基效应同时改变了未占据的MO和被占据的MO。连接基团长度的增加增加了HOMO-2的能量,因此它在观察到的过渡中起着重要作用。简并LUMO(例如克集合通过取代分裂,并且该分裂可通过使用亚丙基去腈基团来增加,该基团显示出最低的能量跃迁和来自母卟啉锌配合物的最大光谱扰动。这些数据得到共振拉曼光谱研究的支持,共振拉曼光谱研究表明,对于高能跃迁,苯基模式显着增强;对于低能跃迁,茚满二酮模式显着增强。










































 京公网安备 11010802027423号
京公网安备 11010802027423号