当前位置:
X-MOL 学术
›
Nano Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Rigorous Method of Calculating Exfoliation Energies from First Principles
Nano Letters ( IF 9.6 ) Pub Date : 2018-04-18 00:00:00 , DOI: 10.1021/acs.nanolett.7b04201 Jong Hyun Jung 1 , Cheol-Hwan Park 1 , Jisoon Ihm 2
Nano Letters ( IF 9.6 ) Pub Date : 2018-04-18 00:00:00 , DOI: 10.1021/acs.nanolett.7b04201 Jong Hyun Jung 1 , Cheol-Hwan Park 1 , Jisoon Ihm 2
Affiliation
|
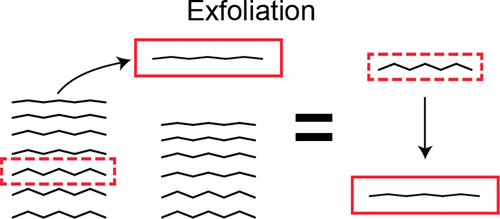
The exfoliation energy, the energy required to peel off an atomic layer from the surface of a bulk material, is of fundamental importance in the science and engineering of two-dimensional materials. Traditionally, the exfoliation energy of a material has been obtained from first-principles by calculating the difference in the ground-state energy between (i) a slab of N atomic layers (N ≫ 1) and (ii) a slab of N – 1 atomic layers plus an atomic layer separated from the slab. In this paper, we prove that the exfoliation energy can be obtained exactly as the difference in the ground-state energy between a bulk material (per atomic layer) and a single isolated layer. The proposed method is (i) tremendously lower in computational cost than the traditional approach because it does not require calculations on thick slabs, (ii) still valid even if there is a surface reconstruction of any kind, (iii) capable of taking into account the relaxation of the single exfoliated layer (both in-plane lattice parameters and atomic positions), and (iv) easily combined with all kinds of many-body computational methods. As a proof of principles, we calculated exfoliation energies of graphene, hexagonal boron nitride, MoS2, and phosphorene using density-functional theory. In addition, we found that the in-plane relaxation of an exfoliated layer accounts for 5% of one-layer exfoliation energy of phosphorene while it is negligible (<0.4%) in the other cases.
中文翻译:

从第一原理计算剥落能量的严格方法
剥落能,即从块状材料表面剥离原子层所需的能量,在二维材料的科学和工程中至关重要。传统上,材料的剥落能是通过计算(i)N个原子层(N≫ 1)的平板与(ii)N – 1的平板之间的基态能量之差而从第一原理获得的原子层加上与平板分离的原子层。在本文中,我们证明了剥离能量可以精确地获得表示散装材料(每个原子层)和单个隔离层之间的基态能量之差。所提出的方法(i)与传统方法相比,其计算成本大大降低,因为它不需要对厚板进行计算;(ii)即使存在任何类型的表面重构,该方法仍然有效;(iii)能够考虑到单个剥离层的松弛(面内晶格参数和原子位置),以及(iv)易于与各种多体计算方法结合使用。作为原理的证明,我们计算了石墨烯,六方氮化硼,MoS 2的剥离能,以及使用密度泛函理论的phosphor。另外,我们发现,剥落层的面内弛豫占磷光体单层剥落能量的5%,而在其他情况下则可忽略不计(<0.4%)。
更新日期:2018-04-18
中文翻译:
从第一原理计算剥落能量的严格方法
剥落能,即从块状材料表面剥离原子层所需的能量,在二维材料的科学和工程中至关重要。传统上,材料的剥落能是通过计算(i)N个原子层(N≫ 1)的平板与(ii)N – 1的平板之间的基态能量之差而从第一原理获得的原子层加上与平板分离的原子层。在本文中,我们证明了剥离能量可以精确地获得表示散装材料(每个原子层)和单个隔离层之间的基态能量之差。所提出的方法(i)与传统方法相比,其计算成本大大降低,因为它不需要对厚板进行计算;(ii)即使存在任何类型的表面重构,该方法仍然有效;(iii)能够考虑到单个剥离层的松弛(面内晶格参数和原子位置),以及(iv)易于与各种多体计算方法结合使用。作为原理的证明,我们计算了石墨烯,六方氮化硼,MoS 2的剥离能,以及使用密度泛函理论的phosphor。另外,我们发现,剥落层的面内弛豫占磷光体单层剥落能量的5%,而在其他情况下则可忽略不计(<0.4%)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号