当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Active learning with non-ab initio input features toward efficient CO2 reduction catalysts†
Chemical Science ( IF 7.6 ) Pub Date : 2018-04-17 00:00:00 , DOI: 10.1039/c7sc03422a Juhwan Noh 1 , Seoin Back 1 , Jaehoon Kim 1 , Yousung Jung 1
Chemical Science ( IF 7.6 ) Pub Date : 2018-04-17 00:00:00 , DOI: 10.1039/c7sc03422a Juhwan Noh 1 , Seoin Back 1 , Jaehoon Kim 1 , Yousung Jung 1
Affiliation
|
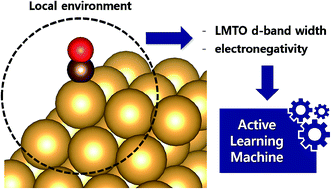
In a conventional chemisorption model, the d-band center theory (augmented sometimes with the upper edge of the d-band for improved accuracy) plays a central role in predicting adsorption energies and catalytic activity as a function of the d-band center of solid surfaces, but it requires density functional calculations that can be quite costly for the purposes of large scale screening of materials. In this work, we propose to use the d-band width of the muffin-tin orbital theory (to account for the local coordination environment) plus electronegativity (to account for adsorbate renormalization) as a simple set of alternative descriptors for chemisorption which do not require ab initio calculations for large-scale first-hand screening. This pair of descriptors is then combined with machine learning methods, namely, neural network (NN) and kernel ridge regression (KRR). We show, for a toy set of 263 alloy systems, that the CO adsorption energy on the (100) facet can be predicted with a mean absolute deviation error of 0.05 eV. We achieved this high accuracy by utilizing an active learning algorithm, without which the accuracy was 0.18 eV. In addition, the results of testing the method with other facets such as (111) terrace and (211) step sites suggest that the present model is also capable of handling different coordination environments effectively. As an example of the practical application of this machine, we identified Cu3Y@Cu* as an active and cost-effective electrochemical CO2 reduction catalyst to produce CO with an overpotential ∼1 V lower than a Au catalyst.
中文翻译:

具有非从头算输入特征的主动学习,以实现高效的 CO2 还原催化剂†
在传统的化学吸附模型中,d 带中心理论(有时会增加 d 带的上边缘以提高准确性)在预测吸附能和催化活性作为固体 d 带中心的函数方面起着核心作用表面,但它需要密度泛函计算,对于大规模筛选材料而言,这可能会非常昂贵。在这项工作中,我们建议使用松饼-锡轨道理论的 d 波段宽度(以说明局部配位环境)加上电负性(以说明吸附质重整化)作为一组简单的化学吸附替代描述符,这些描述符不从头算起用于大规模第一手筛选的计算。然后将这对描述符与机器学习方法相结合,即神经网络(NN)和核岭回归(KRR)。我们表明,对于一组 263 合金系统的玩具,可以预测 (100) 面上的 CO 吸附能,平均绝对偏差误差为 0.05 eV。我们通过使用主动学习算法实现了如此高的准确度,没有它的准确度为 0.18 eV。此外,该方法与其他方面如(111)阶地和(211)台阶点的测试结果表明,本模型也能够有效地处理不同的协调环境。作为该机器实际应用的一个例子,我们确定 Cu 3 Y@Cu* 是一种活性且具有成本效益的电化学 CO2还原催化剂产生CO,其过电势比Au催化剂低~1 V。
更新日期:2018-04-17
中文翻译:
具有非从头算输入特征的主动学习,以实现高效的 CO2 还原催化剂†
在传统的化学吸附模型中,d 带中心理论(有时会增加 d 带的上边缘以提高准确性)在预测吸附能和催化活性作为固体 d 带中心的函数方面起着核心作用表面,但它需要密度泛函计算,对于大规模筛选材料而言,这可能会非常昂贵。在这项工作中,我们建议使用松饼-锡轨道理论的 d 波段宽度(以说明局部配位环境)加上电负性(以说明吸附质重整化)作为一组简单的化学吸附替代描述符,这些描述符不从头算起用于大规模第一手筛选的计算。然后将这对描述符与机器学习方法相结合,即神经网络(NN)和核岭回归(KRR)。我们表明,对于一组 263 合金系统的玩具,可以预测 (100) 面上的 CO 吸附能,平均绝对偏差误差为 0.05 eV。我们通过使用主动学习算法实现了如此高的准确度,没有它的准确度为 0.18 eV。此外,该方法与其他方面如(111)阶地和(211)台阶点的测试结果表明,本模型也能够有效地处理不同的协调环境。作为该机器实际应用的一个例子,我们确定 Cu 3 Y@Cu* 是一种活性且具有成本效益的电化学 CO2还原催化剂产生CO,其过电势比Au催化剂低~1 V。










































 京公网安备 11010802027423号
京公网安备 11010802027423号