当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Why one can expect large rectification in molecular junctions based on alkane monothiols and why rectification is so modest†
Chemical Science ( IF 7.6 ) Pub Date : 2018-04-09 00:00:00 , DOI: 10.1039/c8sc00938d Zuoti Xie 1 , Ioan Bâldea 2 , C Daniel Frisbie 1
Chemical Science ( IF 7.6 ) Pub Date : 2018-04-09 00:00:00 , DOI: 10.1039/c8sc00938d Zuoti Xie 1 , Ioan Bâldea 2 , C Daniel Frisbie 1
Affiliation
|
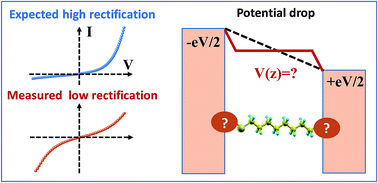
Many attempts to obtain high current rectification ratios (RRs) in molecular electronics are triggered by a potentiometer rule argument, which predicts that a strongly asymmetric location of the dominant molecular orbital yields large RR-values. Invoking this argument, molecular junctions based on alkane monothiols (CnT) can be expected to exhibit high RRs; the HOMO of these molecules is localized on the thiol terminal group bonded to one electrode. The extensive current–voltage (I–V) results for CP-AFM (conducting probe atomic force microscope) CnT junctions of various molecular lengths (n = 7, 8, 9, 10, and 12) and different metallic contacts (Ag, Au, and Pt) are consistent with conduction dominated by the HOMO, but the measured RR ∼ 1.5 is much smaller than that predicted by the potentiometer rule framework. Further, the linear shift in the HOMO position with applied bias, γ, which gives rise to rectification, is also smaller than expected, and critically, γ has the opposite sign from potentiometer rule predictions. Companion ab initio OVGF (outer valence Green's function) quantum chemical calculations provide important insight. Namely, a linear Stark shift γm is calculated for the HOMO of CnT molecules for electric field strengths (106–107 V cm−1) typical of molecular junctions, and the sign of γm matches the sign of the experimental γ for junctions derived from transport measurements, suggesting that the Stark effect plays an important role. However, the magnitude of the measured γ is only 10–15% of the computed value γm. We propose that this implies that the contacts are far from optimal; they substantially screen the effect of the applied bias, possibly via molecule–electrode interface states. We predict that, with optimized contacts, the rectification ratios in CnT-based junctions can reach reasonably high values (RR ≈ 500). We believe that Stark shifts and limited current rectification due to non-ideal contacts discussed here for the specific case of alkane monothiol junctions are issues of general interest for molecular electronics that deserve further consideration.
中文翻译:

为什么人们可以预期基于烷烃一硫醇的分子连接会发生大规模整流,以及为什么整流如此温和†
在分子电子学中获得高电流整流比 (RR) 的许多尝试都是由电位计规则论证引发的,该论证预测主分子轨道的强烈不对称位置会产生大的 RR 值。援引这一论点,基于烷烃单硫醇 (CnT) 的分子连接有望表现出高 RR;这些分子的 HOMO 位于与一个电极键合的硫醇端基上。各种分子长度( n = 7、8、9、10 和 12)和不同金属触点(Ag、Au)的 CP-AFM(传导探针原子力显微镜)CnT 结的广泛电流-电压( I – V ) 结果,和 Pt)与 HOMO 主导的传导一致,但测量的 RR ∼ 1.5 比电位计规则框架预测的要小得多。此外,施加偏置的 HOMO 位置的线性偏移γ(引起整流)也小于预期,并且关键的是,γ具有与电位计规则预测相反的符号。配套的从头算OVGF(外价格林函数)量子化学计算提供了重要的见解。即,对于分子结典型的电场强度(10 6 –10 7 V cm -1 ) ,计算 CnT 分子的 HOMO 的线性斯塔克位移γ m ,并且γ m的符号与实验γ的符号匹配由传输测量得出的连接点,表明斯塔克效应起着重要作用。然而,测量的γ的大小仅为计算值γ m的 10-15% 。我们认为这意味着接触远非最佳;它们可能通过分子-电极界面态基本上屏蔽了施加偏压的影响。我们预测,通过优化接触,基于 CnT 的结中的整流比可以达到相当高的值 (RR ≈ 500)。我们认为,本文针对烷烃单硫醇结的具体情况讨论的非理想接触导致的斯塔克位移和有限电流整流是分子电子学普遍感兴趣的问题,值得进一步考虑。
更新日期:2018-04-09
中文翻译:
为什么人们可以预期基于烷烃一硫醇的分子连接会发生大规模整流,以及为什么整流如此温和†
在分子电子学中获得高电流整流比 (RR) 的许多尝试都是由电位计规则论证引发的,该论证预测主分子轨道的强烈不对称位置会产生大的 RR 值。援引这一论点,基于烷烃单硫醇 (CnT) 的分子连接有望表现出高 RR;这些分子的 HOMO 位于与一个电极键合的硫醇端基上。各种分子长度( n = 7、8、9、10 和 12)和不同金属触点(Ag、Au)的 CP-AFM(传导探针原子力显微镜)CnT 结的广泛电流-电压( I – V ) 结果,和 Pt)与 HOMO 主导的传导一致,但测量的 RR ∼ 1.5 比电位计规则框架预测的要小得多。此外,施加偏置的 HOMO 位置的线性偏移γ(引起整流)也小于预期,并且关键的是,γ具有与电位计规则预测相反的符号。配套的从头算OVGF(外价格林函数)量子化学计算提供了重要的见解。即,对于分子结典型的电场强度(10 6 –10 7 V cm -1 ) ,计算 CnT 分子的 HOMO 的线性斯塔克位移γ m ,并且γ m的符号与实验γ的符号匹配由传输测量得出的连接点,表明斯塔克效应起着重要作用。然而,测量的γ的大小仅为计算值γ m的 10-15% 。我们认为这意味着接触远非最佳;它们可能通过分子-电极界面态基本上屏蔽了施加偏压的影响。我们预测,通过优化接触,基于 CnT 的结中的整流比可以达到相当高的值 (RR ≈ 500)。我们认为,本文针对烷烃单硫醇结的具体情况讨论的非理想接触导致的斯塔克位移和有限电流整流是分子电子学普遍感兴趣的问题,值得进一步考虑。










































 京公网安备 11010802027423号
京公网安备 11010802027423号