当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Proteogenomics of Malignant Melanoma Cell Lines: The Effect of Stringency of Exome Data Filtering on Variant Peptide Identification in Shotgun Proteomics
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-04-16 , DOI: 10.1021/acs.jproteome.7b00841 Anna A. Lobas 1, 2 , Mikhail A. Pyatnitskiy 3, 4 , Alexey L. Chernobrovkin 5 , Irina Y. Ilina 3 , Dmitry S. Karpov 3, 6 , Elizaveta M. Solovyeva 2 , Ksenia G. Kuznetsova 3 , Mark V. Ivanov 2 , Elena Y. Lyssuk 7 , Anna A. Kliuchnikova 3, 8 , Olga E. Voronko 3 , Sergey S. Larin 7 , Roman A. Zubarev 5 , Mikhail V. Gorshkov 2 , Sergei A. Moshkovskii 3, 8
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-04-16 , DOI: 10.1021/acs.jproteome.7b00841 Anna A. Lobas 1, 2 , Mikhail A. Pyatnitskiy 3, 4 , Alexey L. Chernobrovkin 5 , Irina Y. Ilina 3 , Dmitry S. Karpov 3, 6 , Elizaveta M. Solovyeva 2 , Ksenia G. Kuznetsova 3 , Mark V. Ivanov 2 , Elena Y. Lyssuk 7 , Anna A. Kliuchnikova 3, 8 , Olga E. Voronko 3 , Sergey S. Larin 7 , Roman A. Zubarev 5 , Mikhail V. Gorshkov 2 , Sergei A. Moshkovskii 3, 8
Affiliation
|
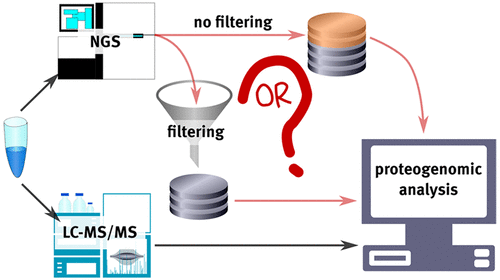
The identification of genetically encoded variants at the proteome level is an important problem in cancer proteogenomics. The generation of customized protein databases from DNA or RNA sequencing data is a crucial stage of the identification workflow. Genomic data filtering applied at this stage may significantly modify variant search results, yet its effect is generally left out of the scope of proteogenomic studies. In this work, we focused on this impact using data of exome sequencing and LC–MS/MS analyses of six replicates for eight melanoma cell lines processed by a proteogenomics workflow. The main objectives were identifying variant peptides and revealing the role of the genomic data filtering in the variant identification. A series of six confidence thresholds for single nucleotide polymorphisms and indels from the exome data were applied to generate customized sequence databases of different stringency. In the searches against unfiltered databases, between 100 and 160 variant peptides were identified for each of the cell lines using X!Tandem and MS-GF+ search engines. The recovery rate for variant peptides was ∼1%, which is approximately three times lower than that of the wild-type peptides. Using unfiltered genomic databases for variant searches resulted in higher sensitivity and selectivity of the proteogenomic workflow and positively affected the ability to distinguish the cell lines based on variant peptide signatures.
中文翻译:

蛋白质组学的恶性黑色素瘤细胞系:Shot弹枪蛋白质组学中外显子组数据过滤的严格性对多肽变异鉴定的影响
在蛋白质组学水平上鉴定遗传编码的变体是癌症蛋白质组学中的重要问题。从DNA或RNA测序数据生成定制的蛋白质数据库是鉴定工作流程的关键阶段。在此阶段应用的基因组数据过滤可能会显着修改变体搜索结果,但其影响通常不在蛋白质组学研究的范围之内。在这项工作中,我们使用蛋白质组学工作流程处理的八种黑色素瘤细胞系的外显子组测序数据和六次重复的LC-MS / MS分析,着眼于这种影响。主要目的是鉴定变体肽,并揭示基因组数据过滤在变体鉴定中的作用。应用来自外显子组数据的单核苷酸多态性和插入缺失的一系列六个置信度阈值来生成不同严格度的定制序列数据库。在针对未过滤数据库的搜索中,使用X!Tandem和MS-GF +搜索引擎为每种细胞系鉴定了100至160种变体肽。变异肽的回收率约为1%,约为野生型肽的三倍。使用未经过滤的基因组数据库进行变体搜索会导致蛋白质组学工作流程的灵敏度和选择性更高,并积极影响基于变体肽标记区分细胞系的能力。使用X!Tandem和MS-GF +搜索引擎为每个细胞系鉴定了100至160个变异肽。变异肽的回收率约为1%,约为野生型肽的三倍。使用未经过滤的基因组数据库进行变体搜索会导致蛋白质组学工作流程的灵敏度和选择性更高,并积极影响基于变体肽标记区分细胞系的能力。使用X!Tandem和MS-GF +搜索引擎为每个细胞系鉴定了100至160个变异肽。变异肽的回收率约为1%,约为野生型肽的三倍。使用未经过滤的基因组数据库进行变体搜索会导致蛋白质组学工作流程的灵敏度和选择性更高,并积极影响基于变体肽标记区分细胞系的能力。
更新日期:2018-04-16
中文翻译:
蛋白质组学的恶性黑色素瘤细胞系:Shot弹枪蛋白质组学中外显子组数据过滤的严格性对多肽变异鉴定的影响
在蛋白质组学水平上鉴定遗传编码的变体是癌症蛋白质组学中的重要问题。从DNA或RNA测序数据生成定制的蛋白质数据库是鉴定工作流程的关键阶段。在此阶段应用的基因组数据过滤可能会显着修改变体搜索结果,但其影响通常不在蛋白质组学研究的范围之内。在这项工作中,我们使用蛋白质组学工作流程处理的八种黑色素瘤细胞系的外显子组测序数据和六次重复的LC-MS / MS分析,着眼于这种影响。主要目的是鉴定变体肽,并揭示基因组数据过滤在变体鉴定中的作用。应用来自外显子组数据的单核苷酸多态性和插入缺失的一系列六个置信度阈值来生成不同严格度的定制序列数据库。在针对未过滤数据库的搜索中,使用X!Tandem和MS-GF +搜索引擎为每种细胞系鉴定了100至160种变体肽。变异肽的回收率约为1%,约为野生型肽的三倍。使用未经过滤的基因组数据库进行变体搜索会导致蛋白质组学工作流程的灵敏度和选择性更高,并积极影响基于变体肽标记区分细胞系的能力。使用X!Tandem和MS-GF +搜索引擎为每个细胞系鉴定了100至160个变异肽。变异肽的回收率约为1%,约为野生型肽的三倍。使用未经过滤的基因组数据库进行变体搜索会导致蛋白质组学工作流程的灵敏度和选择性更高,并积极影响基于变体肽标记区分细胞系的能力。使用X!Tandem和MS-GF +搜索引擎为每个细胞系鉴定了100至160个变异肽。变异肽的回收率约为1%,约为野生型肽的三倍。使用未经过滤的基因组数据库进行变体搜索会导致蛋白质组学工作流程的灵敏度和选择性更高,并积极影响基于变体肽标记区分细胞系的能力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号