当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
In Silico Enhancing M. tuberculosis Protein Interaction Networks in STRING To Predict Drug-Resistance Pathways and Pharmacological Risks
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-04-06 00:00:00 , DOI: 10.1021/acs.jproteome.7b00702 Suyu Mei 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-04-06 00:00:00 , DOI: 10.1021/acs.jproteome.7b00702 Suyu Mei 1
Affiliation
|
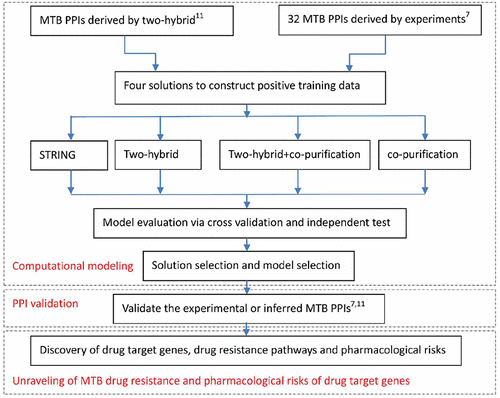
Bacterial protein–protein interaction (PPI) networks are significant to reveal the machinery of signal transduction and drug resistance within bacterial cells. The database STRING has collected a large number of bacterial pathogen PPI networks, but most of the data are of low quality without being experimentally or computationally validated, thus restricting its further biomedical applications. We exploit the experimental data via four solutions to enhance the quality of M. tuberculosis H37Rv (MTB) PPI networks in STRING. Computational results show that the experimental data derived jointly by two-hybrid and copurification approaches are the most reliable to train an L2-regularized logistic regression model for MTB PPI network validation. On the basis of the validated MTB PPI networks, we further study the three problems via breadth-first graph search algorithm: (1) discovery of MTB drug-resistance pathways through searching for the paths between known drug-target genes and drug-resistance genes, (2) choosing potential cotarget genes via searching for the critical genes located on multiple pathways, and (3) choosing essential drug-target genes via analysis of network degree distribution. In addition, we further combine the validated MTB PPI networks with human PPI networks to analyze the potential pharmacological risks of known and candidate drug-target genes from the point of view of system pharmacology. The evidence from protein structure alignment demonstrates that the drugs that act on MTB target genes could also adversely act on human signaling pathways.
中文翻译:

在计算机上增强STRING中的结核分枝杆菌蛋白相互作用网络以预测耐药性途径和药理风险
细菌蛋白间相互作用(PPI)网络对于揭示细菌细胞内信号转导和耐药性具有重要意义。数据库STRING已收集了大量细菌病原体PPI网络,但是大多数数据质量较差,未经实验或计算验证,因此限制了其进一步的生物医学应用。我们通过四种解决方案来利用实验数据来提高STRING中结核分枝杆菌H37Rv(MTB)PPI网络的质量。计算结果表明,通过双杂交和共纯化方法联合获得的实验数据是训练L 2的最可靠方法。-MTB PPI网络验证的规则化Logistic回归模型。在经过验证的MTB PPI网络的基础上,我们通过广度优先图搜索算法进一步研究了三个问题:(1)通过搜索已知的药物靶基因和耐药基因之间的路径来发现MTB耐药路径;(2)通过搜索位于多个途径的关键基因选择潜在的共靶基因,以及(3)通过网络度分布分析选择必需的药物靶基因。此外,我们还将经过验证的MTB PPI网络与人类PPI网络相结合,从系统药理学的角度分析已知和候选药物靶基因的潜在药理学风险。
更新日期:2018-04-07
中文翻译:
在计算机上增强STRING中的结核分枝杆菌蛋白相互作用网络以预测耐药性途径和药理风险
细菌蛋白间相互作用(PPI)网络对于揭示细菌细胞内信号转导和耐药性具有重要意义。数据库STRING已收集了大量细菌病原体PPI网络,但是大多数数据质量较差,未经实验或计算验证,因此限制了其进一步的生物医学应用。我们通过四种解决方案来利用实验数据来提高STRING中结核分枝杆菌H37Rv(MTB)PPI网络的质量。计算结果表明,通过双杂交和共纯化方法联合获得的实验数据是训练L 2的最可靠方法。-MTB PPI网络验证的规则化Logistic回归模型。在经过验证的MTB PPI网络的基础上,我们通过广度优先图搜索算法进一步研究了三个问题:(1)通过搜索已知的药物靶基因和耐药基因之间的路径来发现MTB耐药路径;(2)通过搜索位于多个途径的关键基因选择潜在的共靶基因,以及(3)通过网络度分布分析选择必需的药物靶基因。此外,我们还将经过验证的MTB PPI网络与人类PPI网络相结合,从系统药理学的角度分析已知和候选药物靶基因的潜在药理学风险。










































 京公网安备 11010802027423号
京公网安备 11010802027423号