Journal of Fluorine Chemistry ( IF 1.7 ) Pub Date : 2018-03-31 , DOI: 10.1016/j.jfluchem.2018.03.017 Vahid Saheb , Mahdiyeh Javanmardi
|
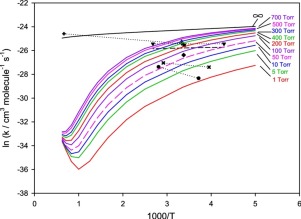
The potential energy surface of the CF3 + O2 reaction is explored by using quantum-chemical combination methods including CBS-QB3, G3B3 and G4. A TST/RRKM model along with a modified strong collision approximation is employed to calculate the thermal rate coefficients for important product channels as a function of temperature and pressure. The calculated results show that the overall rate constant is pressure-dependent over a wide temperature range. At low temperatures, the dominant process is the formation of CF3OO adduct with negative activation energy. However, at higher temperatures, the product channel CF3O + O becomes important. The unimolecular rate coefficients for the thermal decomposition of CF3OO are also computed.
中文翻译:
CF 3自由基与氧分子反应机理和动力学的理论研究
通过使用包括CBS-QB3,G3B3和G4在内的量子化学组合方法探索CF 3 + O 2反应的势能面。使用TST / RRKM模型以及经过修改的强碰撞近似值来计算重要产品通道的热速率系数,该系数是温度和压力的函数。计算结果表明,在较宽的温度范围内,总速率常数与压力有关。在低温下,主要过程是形成具有负活化能的CF 3 OO加合物。但是,在较高温度下,产品通道CF 3 O + O变得很重要。CF 3热分解的单分子速率系数还计算OO。










































 京公网安备 11010802027423号
京公网安备 11010802027423号