Chemical Physics ( IF 2.0 ) Pub Date : 2018-03-17 , DOI: 10.1016/j.chemphys.2018.03.018 Nitin R. Gulvi , Priyanka Patel , Purav M. Badani
|
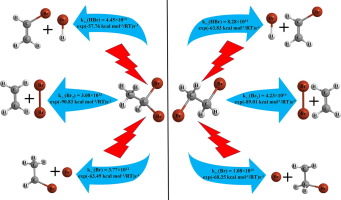
Pathway for dissociation of multihalogenated alkyls is observed to be competitive between molecular and atomic elimination products. Factors such as molecular structure, temperature and pressure are known to influence the same. Hence present work is focussed to explore mechanism and kinetics of atomic (Br) and molecular (HBr and Br2) elimination upon pyrolysis of 1,1– and 1,2–ethyl dibromide (EDB). For this purpose, electronic structure calculations were performed at DFT and CCSD(T) level of theory. In addition to concerted mechanism, an alternate energetically efficient isomerisation pathway has been exploited for molecular elimination. Energy calculations are further complimented by detailed kinetic investigation, over wide range of temperature and pressure, using suitable models like Canonical Transition State Theory, Statistical Adiabatic Channel Model and Troe's formalism. Our calculations suggest high branching ratio for dehydrohalogentation reaction, from both isomers of EDB. Fall off curve depicts good agreement between theoretically estimated and experimentally reported values.
中文翻译:
通过电子结构计算探索二溴乙烷的单分子解离动力学
观察到多卤代烷基解离的途径在分子消除产物和原子消除产物之间具有竞争性。已知诸如分子结构,温度和压力的因素会影响它们。因此,目前的工作重点是探索原子(Br)和分子(HBr和Br 2)的机理和动力学。)在热解1,1–和1,2–乙基二溴化物(EDB)时消除。为此,在DFT和CCSD(T)的理论水平上进行了电子结构计算。除了协同作用的机制外,还开发了另一种能量有效的异构化途径来消除分子。通过使用适当的模型,如标准过渡态理论,统计绝热通道模型和Troe形式主义,在广泛的温度和压力范围内进行详细的动力学研究,进一步补充了能量计算。我们的计算表明,EDB的两个异构体均具有较高的支化比,用于脱卤化氢反应。衰减曲线描述了理论估计值和实验报告值之间的良好一致性。








































 京公网安备 11010802027423号
京公网安备 11010802027423号