当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
MELD-Path Efficiently Computes Conformational Transitions, Including Multiple and Diverse Paths
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-03-16 00:00:00 , DOI: 10.1021/acs.jctc.7b01294 Alberto Perez 1 , Florian Sittel 2 , Gerhard Stock 2 , Ken Dill 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-03-16 00:00:00 , DOI: 10.1021/acs.jctc.7b01294 Alberto Perez 1 , Florian Sittel 2 , Gerhard Stock 2 , Ken Dill 1
Affiliation
|
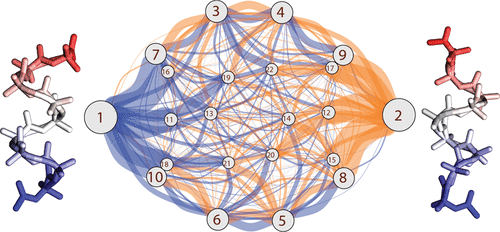
The molecular actions of proteins occur along reaction coordinates. Current computer methods have limited ability to explore them. We describe a fast protocol called MELD-path that (1) efficiently samples relevant conformational states via MELD, an accelerator of Molecular Dynamics (MD), (2) seeds multiple short MD trajectories from MELD states, and then (3) constructs Markov State Models (MSM) that give the routes and kinetics. We tested the method against extensive (multi μs) MD simulations of the right-handed- to left-handed-helix transition of a 9-mer peptide of AIB, the symmetry of which allows us to establish convergence. MELD-path finds all the metastable states, their correct relative populations, and the full ensemble of routes, not just a single assumed route. For this transition, we find a very broad route structure. MELD-path is highly parallelizable and efficient, yielding the full route map in a few days of computation. We believe MELD-path could be a general and rapid way to explore mechanistic processes in biomolecules on the computer.
中文翻译:

MELD-Path 有效计算构象转换,包括多个不同的路径
蛋白质的分子作用沿着反应坐标发生。当前的计算机方法探索它们的能力有限。我们描述了一种称为 MELD-path 的快速协议,它 (1) 通过 MELD、分子动力学 (MD) 的加速器有效地采样相关的构象状态,(2) 从 MELD 状态种子多个短 MD 轨迹,然后 (3) 构造马尔可夫状态提供路线和动力学的模型 (MSM)。我们针对 AIB 的 9 聚体肽的右手向左手螺旋跃迁的广泛(多微秒)MD 模拟测试了该方法,其对称性使我们能够建立收敛。MELD-path 找到所有亚稳态、它们正确的相对种群以及完整的路线集合,而不仅仅是一条假设的路线。对于这种过渡,我们发现了一个非常广泛的路线结构。MELD-path 具有高度并行性和高效性,可在几天的计算中生成完整的路线图。我们相信 MELD-path 可能是一种在计算机上探索生物分子机械过程的通用且快速的方法。
更新日期:2018-03-16
中文翻译:
MELD-Path 有效计算构象转换,包括多个不同的路径
蛋白质的分子作用沿着反应坐标发生。当前的计算机方法探索它们的能力有限。我们描述了一种称为 MELD-path 的快速协议,它 (1) 通过 MELD、分子动力学 (MD) 的加速器有效地采样相关的构象状态,(2) 从 MELD 状态种子多个短 MD 轨迹,然后 (3) 构造马尔可夫状态提供路线和动力学的模型 (MSM)。我们针对 AIB 的 9 聚体肽的右手向左手螺旋跃迁的广泛(多微秒)MD 模拟测试了该方法,其对称性使我们能够建立收敛。MELD-path 找到所有亚稳态、它们正确的相对种群以及完整的路线集合,而不仅仅是一条假设的路线。对于这种过渡,我们发现了一个非常广泛的路线结构。MELD-path 具有高度并行性和高效性,可在几天的计算中生成完整的路线图。我们相信 MELD-path 可能是一种在计算机上探索生物分子机械过程的通用且快速的方法。










































 京公网安备 11010802027423号
京公网安备 11010802027423号