当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fast and flexible gpu accelerated binding free energy calculations within the amber molecular dynamics package
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-12 , DOI: 10.1002/jcc.25187 Daniel J. Mermelstein 1 , Charles Lin 1, 2 , Gard Nelson 3 , Rachael Kretsch 1, 4 , J. Andrew McCammon 1 , Ross C. Walker 1, 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-12 , DOI: 10.1002/jcc.25187 Daniel J. Mermelstein 1 , Charles Lin 1, 2 , Gard Nelson 3 , Rachael Kretsch 1, 4 , J. Andrew McCammon 1 , Ross C. Walker 1, 2
Affiliation
|
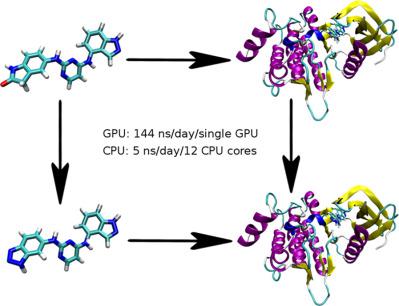
Alchemical free energy (AFE) calculations based on molecular dynamics (MD) simulations are key tools in both improving our understanding of a wide variety of biological processes and accelerating the design and optimization of therapeutics for numerous diseases. Computing power and theory have, however, long been insufficient to enable AFE calculations to be routinely applied in early stage drug discovery. One of the major difficulties in performing AFE calculations is the length of time required for calculations to converge to an ensemble average. CPU implementations of MD‐based free energy algorithms can effectively only reach tens of nanoseconds per day for systems on the order of 50,000 atoms, even running on massively parallel supercomputers. Therefore, converged free energy calculations on large numbers of potential lead compounds are often untenable, preventing researchers from gaining crucial insight into molecular recognition, potential druggability and other crucial areas of interest. Graphics Processing Units (GPUs) can help address this. We present here a seamless GPU implementation, within the PMEMD module of the AMBER molecular dynamics package, of thermodynamic integration (TI) capable of reaching speeds of >140 ns/day for a 44,907‐atom system, with accuracy equivalent to the existing CPU implementation in AMBER. The implementation described here is currently part of the AMBER 18 beta code and will be an integral part of the upcoming version 18 release of AMBER. © 2018 Wiley Periodicals, Inc.
中文翻译:

琥珀色分子动力学包中快速灵活的 GPU 加速结合自由能计算
基于分子动力学 (MD) 模拟的炼金术自由能 (AFE) 计算是提高我们对各种生物过程的理解以及加速设计和优化多种疾病治疗方法的关键工具。然而,计算能力和理论长期以来一直不足以使 AFE 计算常规应用于早期药物发现。执行 AFE 计算的主要困难之一是计算收敛到集合平均值所需的时间长度。对于 50,000 个原子数量级的系统,即使在大规模并行的超级计算机上运行,基于 MD 的自由能算法的 CPU 实现也只能有效地达到每天几十纳秒。所以,大量潜在先导化合物的收敛自由能计算通常是站不住脚的,这阻碍了研究人员获得对分子识别、潜在成药性和其他重要兴趣领域的重要见解。图形处理单元 (GPU) 可以帮助解决这个问题。我们在此展示了在 AMBER 分子动力学包的 PMEMD 模块内的无缝 GPU 实现,热力学积分 (TI) 能够达到 44,907 原子系统的>140 ns/天的速度,其精度相当于现有的 CPU 实现在琥珀色。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 潜在的成药性和其他重要的兴趣领域。图形处理单元 (GPU) 可以帮助解决这个问题。我们在此展示了在 AMBER 分子动力学包的 PMEMD 模块内的无缝 GPU 实现,热力学积分 (TI) 能够达到 44,907 原子系统的>140 ns/天的速度,其精度相当于现有的 CPU 实现在琥珀色。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 潜在的成药性和其他重要的兴趣领域。图形处理单元 (GPU) 可以帮助解决这个问题。我们在此展示了在 AMBER 分子动力学包的 PMEMD 模块内的无缝 GPU 实现,热力学积分 (TI) 能够达到 44,907 原子系统的>140 ns/天的速度,其精度相当于现有的 CPU 实现在琥珀色。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 热力学积分 (TI) 能够达到 44,907 原子系统的速度 > 140 ns/天,精度相当于 AMBER 中现有的 CPU 实现。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 热力学积分 (TI) 能够达到 44,907 原子系统的速度 > 140 ns/天,精度相当于 AMBER 中现有的 CPU 实现。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc.
更新日期:2018-03-12
中文翻译:
琥珀色分子动力学包中快速灵活的 GPU 加速结合自由能计算
基于分子动力学 (MD) 模拟的炼金术自由能 (AFE) 计算是提高我们对各种生物过程的理解以及加速设计和优化多种疾病治疗方法的关键工具。然而,计算能力和理论长期以来一直不足以使 AFE 计算常规应用于早期药物发现。执行 AFE 计算的主要困难之一是计算收敛到集合平均值所需的时间长度。对于 50,000 个原子数量级的系统,即使在大规模并行的超级计算机上运行,基于 MD 的自由能算法的 CPU 实现也只能有效地达到每天几十纳秒。所以,大量潜在先导化合物的收敛自由能计算通常是站不住脚的,这阻碍了研究人员获得对分子识别、潜在成药性和其他重要兴趣领域的重要见解。图形处理单元 (GPU) 可以帮助解决这个问题。我们在此展示了在 AMBER 分子动力学包的 PMEMD 模块内的无缝 GPU 实现,热力学积分 (TI) 能够达到 44,907 原子系统的>140 ns/天的速度,其精度相当于现有的 CPU 实现在琥珀色。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 潜在的成药性和其他重要的兴趣领域。图形处理单元 (GPU) 可以帮助解决这个问题。我们在此展示了在 AMBER 分子动力学包的 PMEMD 模块内的无缝 GPU 实现,热力学积分 (TI) 能够达到 44,907 原子系统的>140 ns/天的速度,其精度相当于现有的 CPU 实现在琥珀色。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 潜在的成药性和其他重要的兴趣领域。图形处理单元 (GPU) 可以帮助解决这个问题。我们在此展示了在 AMBER 分子动力学包的 PMEMD 模块内的无缝 GPU 实现,热力学积分 (TI) 能够达到 44,907 原子系统的>140 ns/天的速度,其精度相当于现有的 CPU 实现在琥珀色。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 热力学积分 (TI) 能够达到 44,907 原子系统的速度 > 140 ns/天,精度相当于 AMBER 中现有的 CPU 实现。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc. 热力学积分 (TI) 能够达到 44,907 原子系统的速度 > 140 ns/天,精度相当于 AMBER 中现有的 CPU 实现。此处描述的实现目前是 AMBER 18 测试版代码的一部分,并将成为即将发布的 AMBER 18 版的组成部分。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号