当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chemical Dynamics Simulations of Thermal Desorption of Protonated Dialanine from a Perfluorinated Self-Assembled Monolayer Surface
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-03-12 00:00:00 , DOI: 10.1021/acs.jpclett.8b00390 Swapnil C. Kohale 1 , Subha Pratihar 1 , William L. Hase 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-03-12 00:00:00 , DOI: 10.1021/acs.jpclett.8b00390 Swapnil C. Kohale 1 , Subha Pratihar 1 , William L. Hase 1
Affiliation
|
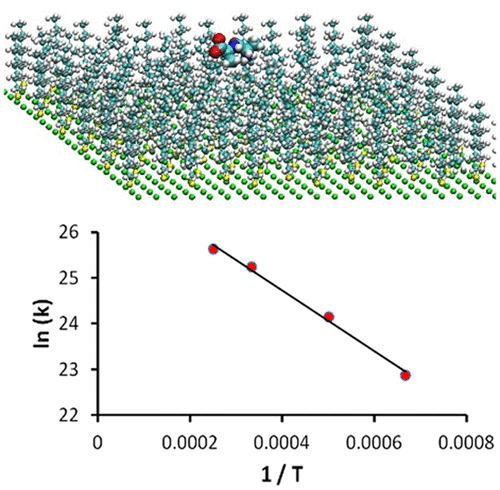
Classical chemical dynamics simulation results are presented for the thermal desorption kinetics and energetics of protonated dialanine ions (ala2-H+) physisorbed on/in a perfluorinated self-assembled monolayer (F-SAM) surface. Previously developed analytic potentials were used for the F-SAM and the ala2-H+/F-SAM intermolecular interaction, and the AMBER valence force field was used for ala2-H+. The activation energy, Ea = 13.2 kcal/mol, determined from the simulations is consistent with previous simulations of the ala2-H+/F-SAM binding energy. The A-factor, 7.8 × 1011 s–1, is about an order of magnitude lower than those representative of small molecule desorption from metal and semiconductor surfaces. This finding is consistent with the decreased entropies of ala2-H+ and the F-SAM upon desorption. Using the Arrhenius parameters for ala2-H+ desorption from the F-SAM, the lifetime of ala2-H+ adsorbed on the F-SAM at 300 K is 5 × 10–3 s. Larger peptide ions are expected to have longer adsorption lifetimes.
中文翻译:

从全氟化自组装单层表面热解吸质子化二氢己胺的化学动力学模拟
针对在全氟化自组装单分子层(F-SAM)表面上物理吸附的质子化二烷离子(ala 2 -H +)的热解吸动力学和能量学,提供了经典的化学动力学模拟结果。先前开发的分析势用于F-SAM和ala 2 -H + / F-SAM的分子间相互作用,而AMBER价力场用于ala 2 -H +。由模拟确定的活化能E a = 13.2 kcal / mol与ala 2 -H + / F-SAM结合能的先前模拟一致。A因子7.8×10 11s –1大约比代表小分子从金属和半导体表面脱附的那些值低一个数量级。这一发现与解吸时ala 2 -H +和F-SAM的熵降低相一致。使用Arrhenius参数ALA 2 -H +从F-SAM解吸,丙氨酸的寿命2 -H +吸附在F-SAM在300 K为5×10 -3秒。较大的肽离子有望具有更长的吸附寿命。
更新日期:2018-03-12
中文翻译:
从全氟化自组装单层表面热解吸质子化二氢己胺的化学动力学模拟
针对在全氟化自组装单分子层(F-SAM)表面上物理吸附的质子化二烷离子(ala 2 -H +)的热解吸动力学和能量学,提供了经典的化学动力学模拟结果。先前开发的分析势用于F-SAM和ala 2 -H + / F-SAM的分子间相互作用,而AMBER价力场用于ala 2 -H +。由模拟确定的活化能E a = 13.2 kcal / mol与ala 2 -H + / F-SAM结合能的先前模拟一致。A因子7.8×10 11s –1大约比代表小分子从金属和半导体表面脱附的那些值低一个数量级。这一发现与解吸时ala 2 -H +和F-SAM的熵降低相一致。使用Arrhenius参数ALA 2 -H +从F-SAM解吸,丙氨酸的寿命2 -H +吸附在F-SAM在300 K为5×10 -3秒。较大的肽离子有望具有更长的吸附寿命。










































 京公网安备 11010802027423号
京公网安备 11010802027423号