Journal of Fluorine Chemistry ( IF 1.7 ) Pub Date : 2018-03-10 , DOI: 10.1016/j.jfluchem.2018.03.001 V. Prakash Reddy , G. K. Surya Prakash , Golam Rasul
|
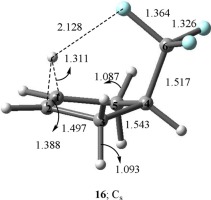
Ab initio calculations at the MP2/cc-pVTZ level of theory show that the classical 1-(trifluoromethyl)cyclopentyl cation (11) is substantially destabilized (by 9.8 kcal/mol) as compared to its isomeric μ-hydrido-bridged global minimum structure 16. However, the classical carbocation 11 is relatively more stable than the nonclassical cyclobutylmethyl cations 6 and 7, by 7.9 and 3.6 kcal/mol, respectively. We have also shown involvement of the noncovalent H⋯F bonding interactions in the μ-hydrido bridged structures, 13, 16, 19, and 20. The latter H⋯F bond distances are dependent on the number of fluorines in the fluoroalkyl group, and decrease in the order: 16 (CF3) > 20 (CHF2) > 19 (CH2F), in accordance with the decreased basicity of the fluorine atom across this series.
中文翻译:
(三氟甲基)环戊基碳正离子的从头算13 C NMR和结构研究
从理论上讲在MP2 / cc-pVTZ水平上的从头算计算表明,与经典的1-(三氟甲基)环戊基阳离子(11)相比,其同分异构的μ-氢桥联全局最小结构大为不稳定(降低了9.8 kcal / mol)。16。然而,经典碳阳离子11比非经典环丁基甲基阳离子6和7相对稳定,分别为7.9kcal / mol和3.6kcal / mol。我们还显示非共价ħ⋯˚F合相互作用参与在μ-氢基桥连的结构,13,16,19,和20。后面的H⋯F键距取决于氟代烷基中氟的数量,并且按照降低的碱度依次降低:16(CF 3)> 20(CHF 2)> 19(CH 2 F)。整个系列中的氟原子。










































 京公网安备 11010802027423号
京公网安备 11010802027423号