Journal of the Taiwan Institute of Chemical Engineers ( IF 5.5 ) Pub Date : 2018-03-09 , DOI: 10.1016/j.jtice.2018.02.026 Meng-huai Han , Chi-cheng Chiu
|
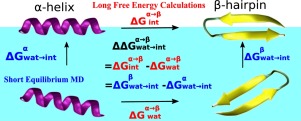
Understanding the protein structural stability and thus its functional integrity at interface are critical for its biotechnology application development. Conventionally, it requires two extensive free energy calculations of protein in bulk and at interface to evaluate the conformational preference change during the adsorption. In this work, we derive an estimation of adsorption free energy at air/water interface for a protein in a defined conformation with the contributions of partial desolvation ΔGdesolv and surface energy ΔGa/w, which can be quickly evaluated from equilibrium MD trajectories. Via thermodynamics cycle, the free energy variation of α-helix to β-hairpin transition during the adsorption can be obtained from the difference between the adsorption free energies of the two conformations. Applying the method, we estimate of 6.31kJ/mol for DP5, in excellent agreement with the MD free energy data of 6.35 kJ/mol. Consistent results for four other tested proteins compared with MD free energy calculation further validate the derived adsorption model. The combination of MD simulations and thermodynamic estimations provide valuable physical insights for protein interfacial folding behaviors and serve as basis for developing prediction tools of protein conformations at interface.
中文翻译:
通过分子动力学模拟快速估算空气/水界面处的蛋白质构象偏好
理解蛋白质的结构稳定性,从而了解其在界面上的功能完整性对于其生物技术应用开发至关重要。常规上,它需要对蛋白质的整体和界面进行两次广泛的自由能计算,以评估吸附过程中构象偏好的变化。在这项工作中,我们得出在空气/水界面吸附自由能的估计的蛋白质与部分去溶剂化的贡献所定义的构象ΔG desolv和表面能ΔG一个/瓦特,可以从平衡MD轨迹快速地评估。通过热力学循环,α-螺旋的自由能变化为β吸附过程中的发夹转变 可以从两个构型的吸附自由能之差获得。应用该方法,我们估计DP5的最大能量为6.31kJ / mol,与MD的6.35 kJ / mol的自由能数据非常吻合。与MD自由能计算相比,其他四种测试蛋白质的一致结果进一步验证了推导的吸附模型。MD模拟和热力学估计的结合为蛋白质界面折叠行为提供了宝贵的物理见解,并为开发界面蛋白质构象的预测工具提供了基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号