当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Characterization of the structural ensembles of p53 TAD2 by molecular dynamics simulations with different force fields†
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-03-06 00:00:00 , DOI: 10.1039/c8cp00067k Yanhua Ouyang 1, 2, 3 , Likun Zhao 1, 2, 3 , Zhuqing Zhang 1, 2, 3
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-03-06 00:00:00 , DOI: 10.1039/c8cp00067k Yanhua Ouyang 1, 2, 3 , Likun Zhao 1, 2, 3 , Zhuqing Zhang 1, 2, 3
Affiliation
|
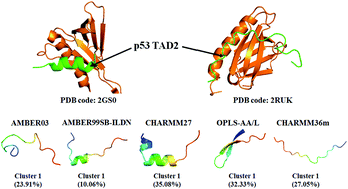
Intrinsically disordered regions (IDRs) or proteins (IDPs), which play crucial biological functions in essential biological processes of life, do not have well-defined secondary or tertiary structures when isolated in solution. The highly dynamic properties and conformational heterogeneity of IDPs make them challenging to study with traditional experimental techniques. As a powerful complementary tool for experiments, all-atom molecular dynamics simulation can obtain detailed conformational information on IDPs, but the limitation of force field accuracy is a challenge for reproducing IDP conformers. Here, we compared five empirical all-atom force fields AMBER03, AMBER99SB-ILDN, CHARMM27, OPLS-AA/L and CHARMM36m in modeling the conformational ensembles of wild-type peptide TAD2(41–62) from the human p53 tumor suppressor. Our results show that for the model peptide, the newest force field CHARMM36m produces more expanded coil ensemble followed by AMBER99SB-ILDN; CHARMM27 displays a predominant propensity for a helical structure; whereas OPLS-AA/L exhibits a apparent preference for a β-sheet structure and yields the most compact conformation. In the comparison of the simulated dimensions with theoretical prediction and the back-calculated chemical shifts with experimental measurements, AMBER99SB-ILDN gives a more consistent agreement than the other force fields. In addition, the region from residues 47 to 55, which commonly forms an amphipathic α-helix upon binding target proteins according to experimental observation, could form a helical structure with a different probability population in our simulations with different force fields. This implies that the binding process might be conducted by, or partly by “conformation selection” for this peptide. This work indicates that force field development for modeling general IDPs accurately has a long way to go, and more detailed experimental data of IDPs are also in demand.
中文翻译:

通过不同力场的分子动力学模拟表征p53 TAD2的结构体†
固有无序区(IDR)或蛋白质(IDP)在生命的基本生物学过程中起着至关重要的生物学功能,当在溶液中分离时,它们没有明确定义的二级或三级结构。IDP的高度动态特性和构象异质性使其很难用传统的实验技术进行研究。作为实验的有力补充工具,全原子分子动力学模拟可以获取有关IDP的详细构象信息,但是力场精度的限制是复制IDP构象异构体的一个挑战。在这里,我们比较了五个经验性全原子力场AMBER03,AMBER99SB-ILDN,CHARMM27,OPLS-AA / L和CHARMM36m,以模拟来自人类p53肿瘤抑制子的野生型肽TAD2(41-62)的构象集合。我们的结果表明,对于模型肽,最新的力场CHARMM36m产生了更多的扩展线圈组,其次是AMBER99SB-ILDN。CHARMM27显示出螺旋结构的主要倾向;而OPLS-AA / L表现出对β-折叠结构的明显偏爱,并产生最紧密的构象。在将模拟尺寸与理论预测值进行比较以及将反算出的化学位移与实验测量值进行比较时,AMBER99SB-ILDN比其他力场具有更一致的一致性。此外,根据实验观察,从残基47到55的区域通常会在结合目标蛋白时形成两亲性α-螺旋,在我们用不同力场进行的模拟中,该区域可能形成具有不同概率群体的螺旋结构。这意味着结合过程可以通过或部分通过该肽的“构象选择”来进行。这项工作表明,准确建模通用IDP的力场开发还有很长的路要走,并且还需要更详细的IDP实验数据。
更新日期:2018-03-06
中文翻译:
通过不同力场的分子动力学模拟表征p53 TAD2的结构体†
固有无序区(IDR)或蛋白质(IDP)在生命的基本生物学过程中起着至关重要的生物学功能,当在溶液中分离时,它们没有明确定义的二级或三级结构。IDP的高度动态特性和构象异质性使其很难用传统的实验技术进行研究。作为实验的有力补充工具,全原子分子动力学模拟可以获取有关IDP的详细构象信息,但是力场精度的限制是复制IDP构象异构体的一个挑战。在这里,我们比较了五个经验性全原子力场AMBER03,AMBER99SB-ILDN,CHARMM27,OPLS-AA / L和CHARMM36m,以模拟来自人类p53肿瘤抑制子的野生型肽TAD2(41-62)的构象集合。我们的结果表明,对于模型肽,最新的力场CHARMM36m产生了更多的扩展线圈组,其次是AMBER99SB-ILDN。CHARMM27显示出螺旋结构的主要倾向;而OPLS-AA / L表现出对β-折叠结构的明显偏爱,并产生最紧密的构象。在将模拟尺寸与理论预测值进行比较以及将反算出的化学位移与实验测量值进行比较时,AMBER99SB-ILDN比其他力场具有更一致的一致性。此外,根据实验观察,从残基47到55的区域通常会在结合目标蛋白时形成两亲性α-螺旋,在我们用不同力场进行的模拟中,该区域可能形成具有不同概率群体的螺旋结构。这意味着结合过程可以通过或部分通过该肽的“构象选择”来进行。这项工作表明,准确建模通用IDP的力场开发还有很长的路要走,并且还需要更详细的IDP实验数据。









































 京公网安备 11010802027423号
京公网安备 11010802027423号