当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Evaluating excited state atomic polarizabilities of chromophores†
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-03-06 00:00:00 , DOI: 10.1039/c7cp08549d Esther Heid 1 , Patricia A Hunt , Christian Schröder
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-03-06 00:00:00 , DOI: 10.1039/c7cp08549d Esther Heid 1 , Patricia A Hunt , Christian Schröder
Affiliation
|
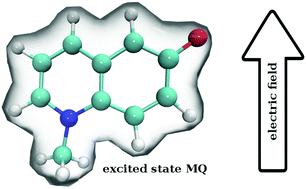
Ground and excited state dipoles and polarizabilities of the chromophores N-methyl-6-oxyquinolinium betaine (MQ) and coumarin 153 (C153) in solution have been evaluated using time-dependent density functional theory (TD-DFT). A method for determining the atomic polarizabilities has been developed; the molecular dipole has been decomposed into atomic charge transfer and polarizability terms, and variation in the presence of an electric field has been used to evaluate atomic polarizabilities. On excitation, MQ undergoes very site-specific changes in polarizability while C153 shows significantly less variation. We also conclude that MQ cannot be adequately described by standard atomic polarizabilities based on atomic number and hybridization state. Changes in the molecular polarizability of MQ (on excitation) are not representative of the local site-specific changes in atomic polarizability, thus the overall molecular polarizability ratio 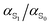 does not provide a good approximation for local atom-specific polarizability changes on excitation. Accurate excited state force fields are needed for computer simulation of solvation dynamics. The chromophores considered in this study are often used as molecular probes. The methods and data reported here can be used for the construction of polarizable ground and excited state force fields. Atomic and molecular polarizabilities (ground and excited states) have been evaluated over a range of functionals and basis sets. Different mechanisms for including solvation effects have been examined; using a polarizable continuum model, explicit solvation and via sampling of clusters extracted from a MD simulation. A range of different solvents have also been considered.
does not provide a good approximation for local atom-specific polarizability changes on excitation. Accurate excited state force fields are needed for computer simulation of solvation dynamics. The chromophores considered in this study are often used as molecular probes. The methods and data reported here can be used for the construction of polarizable ground and excited state force fields. Atomic and molecular polarizabilities (ground and excited states) have been evaluated over a range of functionals and basis sets. Different mechanisms for including solvation effects have been examined; using a polarizable continuum model, explicit solvation and via sampling of clusters extracted from a MD simulation. A range of different solvents have also been considered.
中文翻译:

评估发色团的激发态原子极化率†
使用时间依赖性密度泛函理论 (TD-DFT) 评估了溶液中发色团N-甲基-6-羟基喹啉甜菜碱 (MQ) 和香豆素 153 (C153) 的基态和激发态偶极子以及极化率。已开发出一种测定原子极化率的方法;分子偶极子已分解为原子电荷转移和极化率项,并且电场存在下的变化已用于评估原子极化率。在激发时,MQ 的极化率会发生非常特定位点的变化,而 C153 的变化则明显较小。我们还得出结论,MQ 不能通过基于原子序数和杂化状态的标准原子极化率来充分描述。 MQ 分子极化率的变化(激发时)并不代表原子极化率的局部特定位置变化,因此总体分子极化率 不能为激发时局部原子特定极化率变化提供良好的近似。溶剂化动力学的计算机模拟需要精确的激发态力场。本研究中考虑的发色团通常用作分子探针。这里报告的方法和数据可用于构建可极化基态和激发态力场。原子和分子极化率(基态和激发态)已在一系列泛函和基组上进行了评估。已经检查了包含溶剂化效应的不同机制;使用可极化连续介质模型、显式溶剂化以及通过从 MD 模拟中提取的簇采样。还考虑了一系列不同的溶剂。
更新日期:2018-03-06
does not provide a good approximation for local atom-specific polarizability changes on excitation. Accurate excited state force fields are needed for computer simulation of solvation dynamics. The chromophores considered in this study are often used as molecular probes. The methods and data reported here can be used for the construction of polarizable ground and excited state force fields. Atomic and molecular polarizabilities (ground and excited states) have been evaluated over a range of functionals and basis sets. Different mechanisms for including solvation effects have been examined; using a polarizable continuum model, explicit solvation and via sampling of clusters extracted from a MD simulation. A range of different solvents have also been considered.
中文翻译:
评估发色团的激发态原子极化率†
使用时间依赖性密度泛函理论 (TD-DFT) 评估了溶液中发色团N-甲基-6-羟基喹啉甜菜碱 (MQ) 和香豆素 153 (C153) 的基态和激发态偶极子以及极化率。已开发出一种测定原子极化率的方法;分子偶极子已分解为原子电荷转移和极化率项,并且电场存在下的变化已用于评估原子极化率。在激发时,MQ 的极化率会发生非常特定位点的变化,而 C153 的变化则明显较小。我们还得出结论,MQ 不能通过基于原子序数和杂化状态的标准原子极化率来充分描述。 MQ 分子极化率的变化(激发时)并不代表原子极化率的局部特定位置变化,因此总体分子极化率
不能为激发时局部原子特定极化率变化提供良好的近似。溶剂化动力学的计算机模拟需要精确的激发态力场。本研究中考虑的发色团通常用作分子探针。这里报告的方法和数据可用于构建可极化基态和激发态力场。原子和分子极化率(基态和激发态)已在一系列泛函和基组上进行了评估。已经检查了包含溶剂化效应的不同机制;使用可极化连续介质模型、显式溶剂化以及通过从 MD 模拟中提取的簇采样。还考虑了一系列不同的溶剂。








































 京公网安备 11010802027423号
京公网安备 11010802027423号