当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate lattice energies of organic molecular crystals from periodic turbomole calculations
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-05 , DOI: 10.1002/jcc.25205 Hannes Konrad Buchholz 1, 2 , Matthias Stein 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-05 , DOI: 10.1002/jcc.25205 Hannes Konrad Buchholz 1, 2 , Matthias Stein 2
Affiliation
|
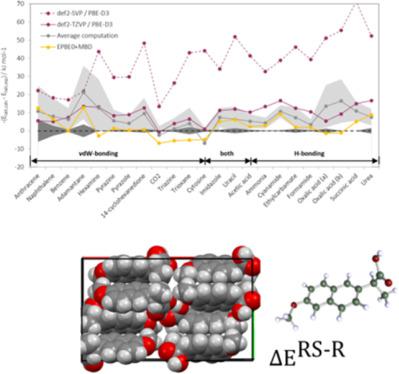
Accurate lattice energies of organic crystals are important i.e. for the pharmaceutical industry. Periodic DFT calculations with atom‐centered Gaussian basis functions with the Turbomole program are used to calculate lattice energies for several non‐covalently bound organic molecular crystals. The accuracy and convergence of results with basis set size and k‐space sampling from periodic calculations is evaluated for the two reference molecules benzoic acid and naphthalene. For the X23 benchmark set of small molecular crystals accurate lattice energies are obtained using the PBE‐D3 functional. In particular for hydrogen‐bonded systems, a sufficiently large basis set is required. The calculated lattice energy differences between enantiopure and racemic crystal forms for a prototype set of chiral molecules are in good agreement with experimental results and allow the rationalization and computer‐aided design of chiral separation processes. © 2018 Wiley Periodicals, Inc.
中文翻译:

来自周期性涡轮分子计算的有机分子晶体的精确晶格能量
有机晶体的准确晶格能很重要,即对于制药工业。使用 Turbomole 程序使用以原子为中心的高斯基函数进行周期性 DFT 计算,用于计算几种非共价结合的有机分子晶体的晶格能量。评估了两个参考分子苯甲酸和萘的基组大小和 k 空间采样的结果的准确性和收敛性。对于 X23 基准小分子晶体集,使用 PBE-D3 泛函获得准确的晶格能量。特别是对于氢键系统,需要足够大的基组。手性分子原型组的对映体纯和外消旋晶型之间计算的晶格能差异与实验结果非常一致,并允许手性分离过程的合理化和计算机辅助设计。© 2018 Wiley Periodicals, Inc.
更新日期:2018-03-05
中文翻译:
来自周期性涡轮分子计算的有机分子晶体的精确晶格能量
有机晶体的准确晶格能很重要,即对于制药工业。使用 Turbomole 程序使用以原子为中心的高斯基函数进行周期性 DFT 计算,用于计算几种非共价结合的有机分子晶体的晶格能量。评估了两个参考分子苯甲酸和萘的基组大小和 k 空间采样的结果的准确性和收敛性。对于 X23 基准小分子晶体集,使用 PBE-D3 泛函获得准确的晶格能量。特别是对于氢键系统,需要足够大的基组。手性分子原型组的对映体纯和外消旋晶型之间计算的晶格能差异与实验结果非常一致,并允许手性分离过程的合理化和计算机辅助设计。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号