当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab initio potential energy surface and vibration-rotation energy levels of germanium dicarbide, GeC2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-05 , DOI: 10.1002/jcc.25204 Jacek Koput 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-05 , DOI: 10.1002/jcc.25204 Jacek Koput 1
Affiliation
|
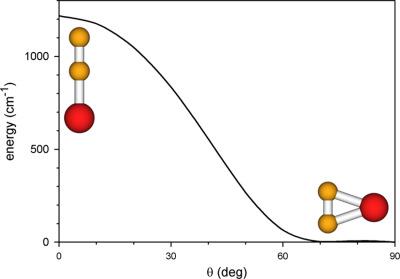
The accurate ground‐state potential energy surface of germanium dicarbide, GeC2, has been determined from ab initio calculations using the coupled‐cluster approach. The core–electron correlation, higher‐order valence‐electron correlation, and scalar relativistic effects were taken into account. The potential energy surface of GeC2 was shown to be extraordinarily flat near the T‐shaped equilibrium configuration. The potential energy barrier to the linear CCGe configuration was predicted to be 1218 cm−1. The vibration–rotation energy levels of some GeC2 isotopologues were calculated using a variational method. The vibrational bending mode ν3 was found to be highly anharmonic, with the fundamental wavenumber being only 58 cm−1. Vibrational progressions due to this mode were predicted for the v1=1 , v2=1 , and v2=2 states of GeC2. © 2018 Wiley Periodicals, Inc.
中文翻译:

二碳化锗 GeC2 从头算势能面和振动-旋转能级
二碳化锗的准确基态势能面 GeC2 已使用耦合簇方法从 ab initio 计算确定。考虑了核电子相关性、高阶价电子相关性和标量相对论效应。GeC2 的势能面在 T 形平衡配置附近非常平坦。预测线性 CCGe 配置的势能势垒为 1218 cm-1。使用变分方法计算了一些 GeC2 同位素体的振动-旋转能级。发现振动弯曲模式 ν3 是高度非谐的,基波数仅为 58 cm-1。对于 GeC2 的 v1=1 、v2=1 和 v2=2 状态,预测了由于这种模式导致的振动进程。
更新日期:2018-03-05
中文翻译:
二碳化锗 GeC2 从头算势能面和振动-旋转能级
二碳化锗的准确基态势能面 GeC2 已使用耦合簇方法从 ab initio 计算确定。考虑了核电子相关性、高阶价电子相关性和标量相对论效应。GeC2 的势能面在 T 形平衡配置附近非常平坦。预测线性 CCGe 配置的势能势垒为 1218 cm-1。使用变分方法计算了一些 GeC2 同位素体的振动-旋转能级。发现振动弯曲模式 ν3 是高度非谐的,基波数仅为 58 cm-1。对于 GeC2 的 v1=1 、v2=1 和 v2=2 状态,预测了由于这种模式导致的振动进程。










































 京公网安备 11010802027423号
京公网安备 11010802027423号